 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInterpreting Manifolds and Graph Neural Embeddings from Internet of Things Traffic Flows
Feb 05, 2026The rapid expansion of Internet of Things (IoT) ecosystems has led to increasingly complex and heterogeneous network topologies. Traditional network monitoring and visualization tools rely on aggregated metrics or static representations, which fail to capture the evolving relationships and structural dependencies between devices. Although Graph Neural Networks (GNNs) offer a powerful way to learn from relational data, their internal representations often remain opaque and difficult to interpret for security-critical operations. Consequently, this work introduces an interpretable pipeline that generates directly visualizable low-dimensional representations by mapping high-dimensional embeddings onto a latent manifold. This projection enables the interpretable monitoring and interoperability of evolving network states, while integrated feature attribution techniques decode the specific characteristics shaping the manifold structure. The framework achieves a classification F1-score of 0.830 for intrusion detection while also highlighting phenomena such as concept drift. Ultimately, the presented approach bridges the gap between high-dimensional GNN embeddings and human-understandable network behavior, offering new insights for network administrators and security analysts.
RNA-KG: An ontology-based knowledge graph for representing interactions involving RNA molecules
Nov 30, 2023The "RNA world" represents a novel frontier for the study of fundamental biological processes and human diseases and is paving the way for the development of new drugs tailored to the patient's biomolecular characteristics. Although scientific data about coding and non-coding RNA molecules are continuously produced and available from public repositories, they are scattered across different databases and a centralized, uniform, and semantically consistent representation of the "RNA world" is still lacking. We propose RNA-KG, a knowledge graph encompassing biological knowledge about RNAs gathered from more than 50 public databases, integrating functional relationships with genes, proteins, and chemicals and ontologically grounded biomedical concepts. To develop RNA-KG, we first identified, pre-processed, and characterized each data source; next, we built a meta-graph that provides an ontological description of the KG by representing all the bio-molecular entities and medical concepts of interest in this domain, as well as the types of interactions connecting them. Finally, we leveraged an instance-based semantically abstracted knowledge model to specify the ontological alignment according to which RNA-KG was generated. RNA-KG can be downloaded in different formats and also queried by a SPARQL endpoint. A thorough topological analysis of the resulting heterogeneous graph provides further insights into the characteristics of the "RNA world". RNA-KG can be both directly explored and visualized, and/or analyzed by applying computational methods to infer bio-medical knowledge from its heterogeneous nodes and edges. The resource can be easily updated with new experimental data, and specific views of the overall KG can be extracted according to the bio-medical problem to be studied.
An Open-Source Knowledge Graph Ecosystem for the Life Sciences
Jul 11, 2023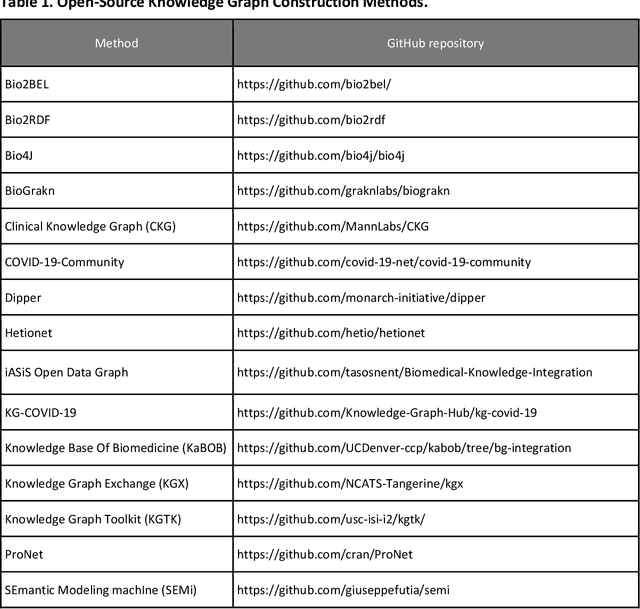
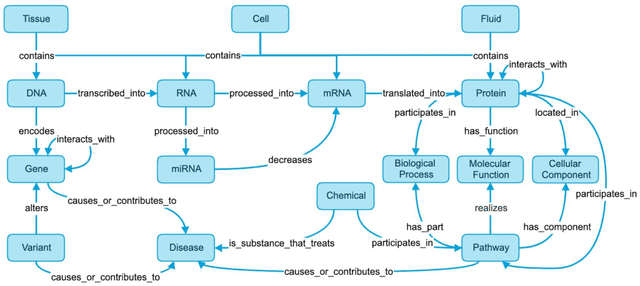
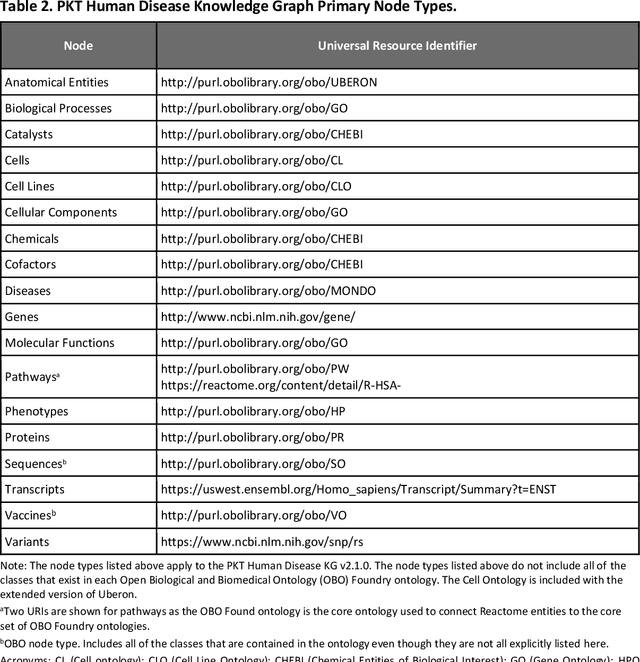
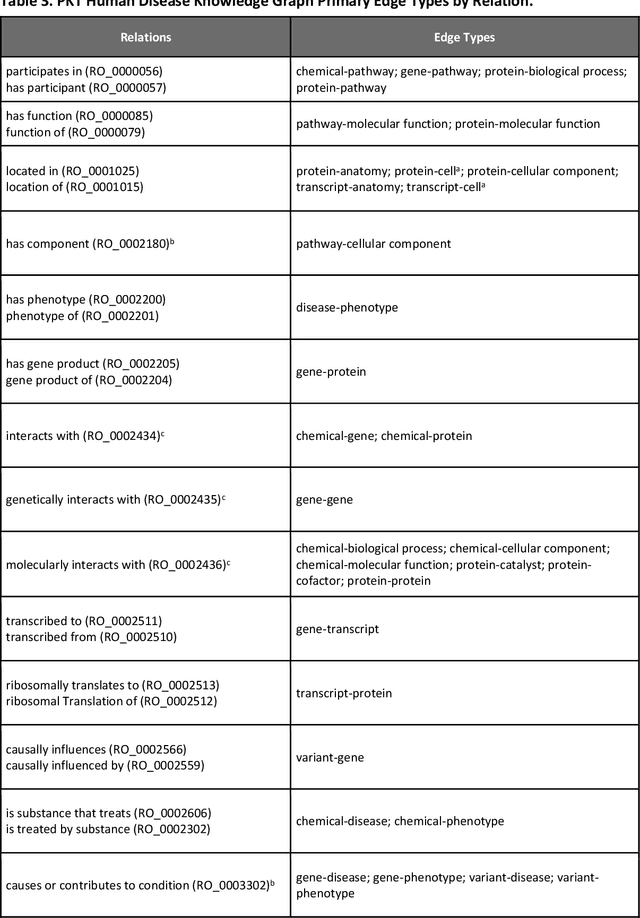
Translational research requires data at multiple scales of biological organization. Advancements in sequencing and multi-omics technologies have increased the availability of these data but researchers face significant integration challenges. Knowledge graphs (KGs) are used to model complex phenomena, and methods exist to automatically construct them. However, tackling complex biomedical integration problems requires flexibility in the way knowledge is modeled. Moreover, existing KG construction methods provide robust tooling at the cost of fixed or limited choices among knowledge representation models. PheKnowLator (Phenotype Knowledge Translator) is a semantic ecosystem for automating the FAIR (Findable, Accessible, Interoperable, and Reusable) construction of ontologically grounded KGs with fully customizable knowledge representation. The ecosystem includes KG construction resources (e.g., data preparation APIs), analysis tools (e.g., SPARQL endpoints and abstraction algorithms), and benchmarks (e.g., prebuilt KGs and embeddings). We evaluate the ecosystem by surveying open-source KG construction methods and analyzing its computational performance when constructing 12 large-scale KGs. With flexible knowledge representation, PheKnowLator enables fully customizable KGs without compromising performance or usability.
A Methodological Framework for the Comparative Evaluation of Multiple Imputation Methods: Multiple Imputation of Race, Ethnicity and Body Mass Index in the U.S. National COVID Cohort Collaborative
Jun 13, 2022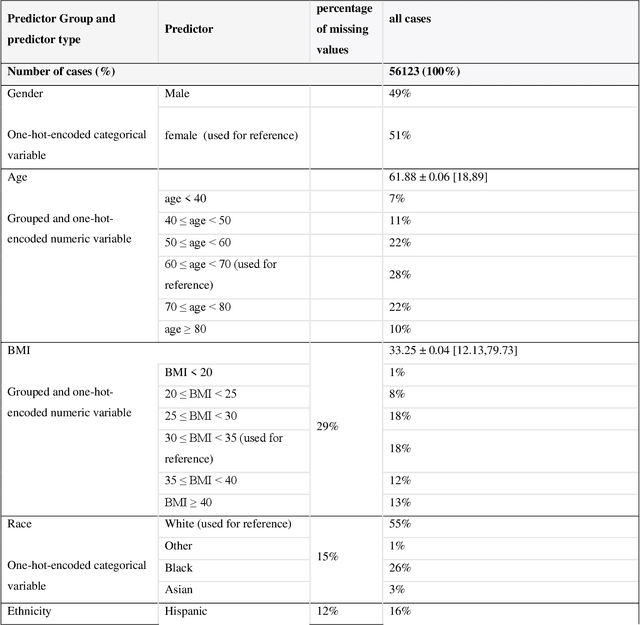
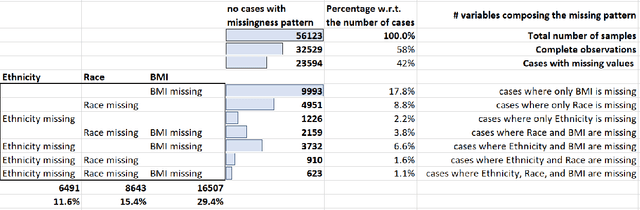

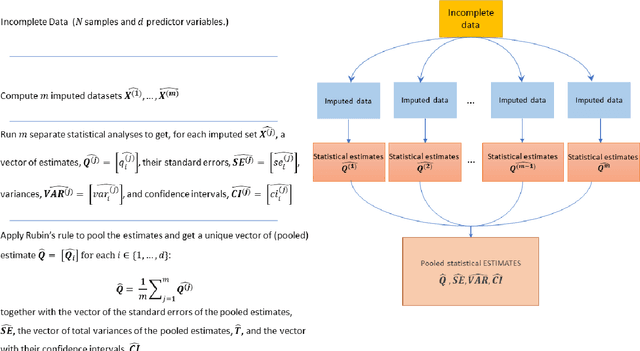
While electronic health records are a rich data source for biomedical research, these systems are not implemented uniformly across healthcare settings and significant data may be missing due to healthcare fragmentation and lack of interoperability between siloed electronic health records. Considering that the deletion of cases with missing data may introduce severe bias in the subsequent analysis, several authors prefer applying a multiple imputation strategy to recover the missing information. Unfortunately, although several literature works have documented promising results by using any of the different multiple imputation algorithms that are now freely available for research, there is no consensus on which MI algorithm works best. Beside the choice of the MI strategy, the choice of the imputation algorithm and its application settings are also both crucial and challenging. In this paper, inspired by the seminal works of Rubin and van Buuren, we propose a methodological framework that may be applied to evaluate and compare several multiple imputation techniques, with the aim to choose the most valid for computing inferences in a clinical research work. Our framework has been applied to validate, and extend on a larger cohort, the results we presented in a previous literature study, where we evaluated the influence of crucial patients' descriptors and COVID-19 severity in patients with type 2 diabetes mellitus whose data is provided by the National COVID Cohort Collaborative Enclave.
GraPE: fast and scalable Graph Processing and Embedding
Oct 12, 2021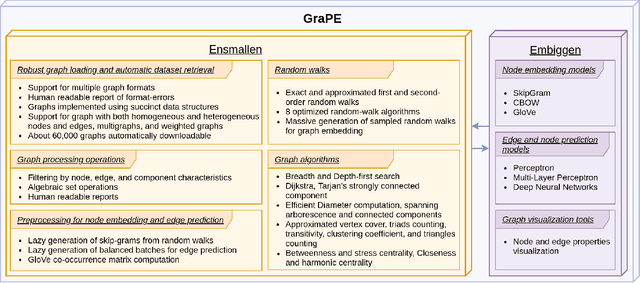
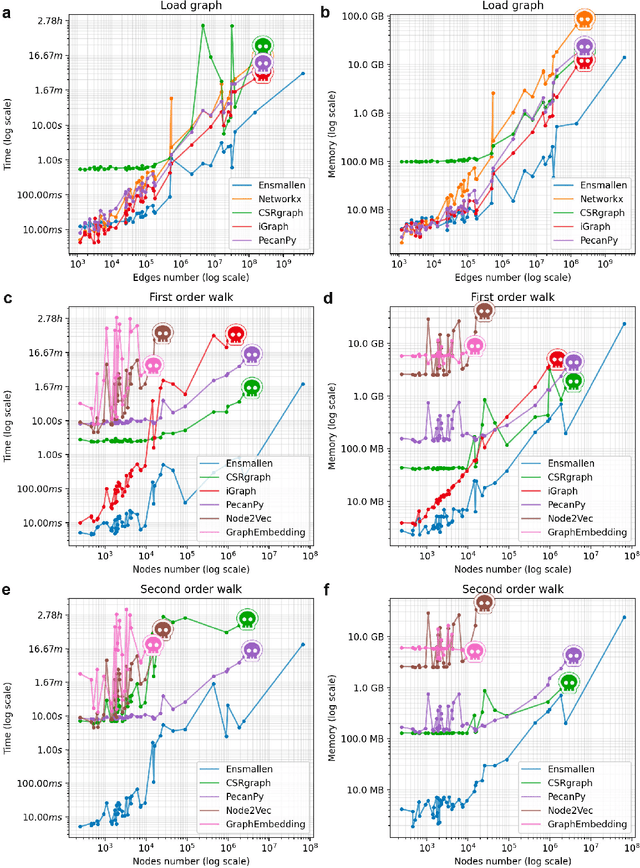
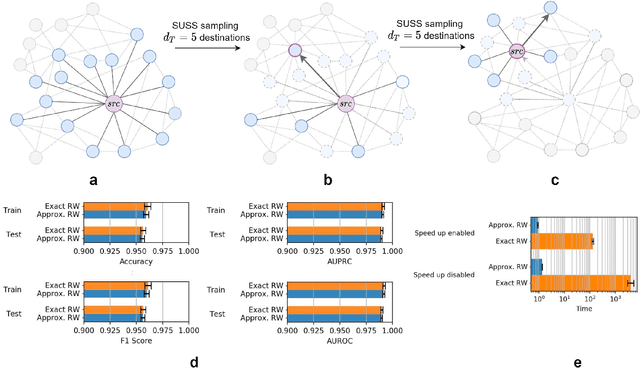
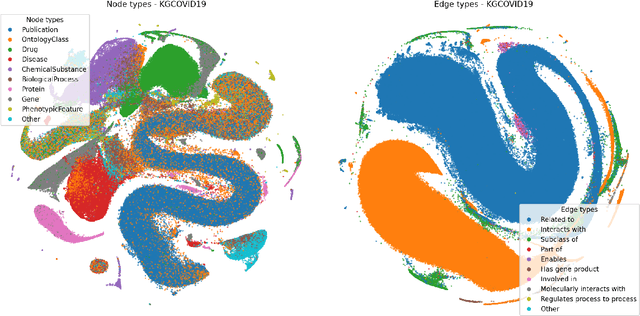
Graph Representation Learning methods have enabled a wide range of learning problems to be addressed for data that can be represented in graph form. Nevertheless, several real world problems in economy, biology, medicine and other fields raised relevant scaling problems with existing methods and their software implementation, due to the size of real world graphs characterized by millions of nodes and billions of edges. We present GraPE, a software resource for graph processing and random walk based embedding, that can scale with large and high-degree graphs and significantly speed up-computation. GraPE comprises specialized data structures, algorithms, and a fast parallel implementation that displays everal orders of magnitude improvement in empirical space and time complexity compared to state of the art software resources, with a corresponding boost in the performance of machine learning methods for edge and node label prediction and for the unsupervised analysis of graphs.GraPE is designed to run on laptop and desktop computers, as well as on high performance computing clusters
Het-node2vec: second order random walk sampling for heterogeneous multigraphs embedding
Jan 05, 2021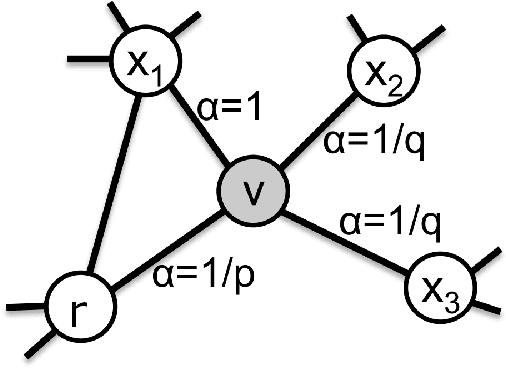
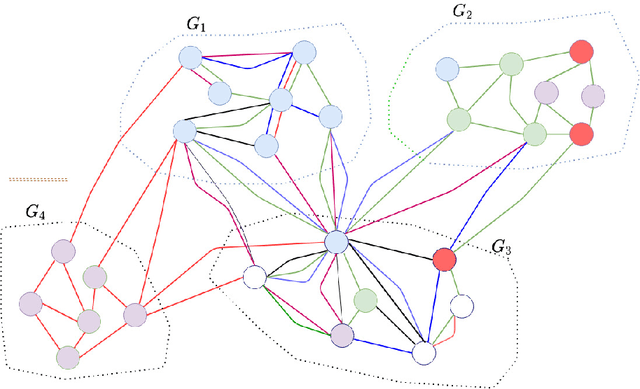

We introduce a set of algorithms (Het-node2vec) that extend the original node2vec node-neighborhood sampling method to heterogeneous multigraphs, i.e. networks characterized by multiple types of nodes and edges. The resulting random walk samples capture both the structural characteristics of the graph and the semantics of the different types of nodes and edges. The proposed algorithms can focus their attention on specific node or edge types, allowing accurate representations also for underrepresented types of nodes/edges that are of interest for the prediction problem under investigation. These rich and well-focused representations can boost unsupervised and supervised learning on heterogeneous graphs.
Multitask Hopfield Networks
Apr 10, 2019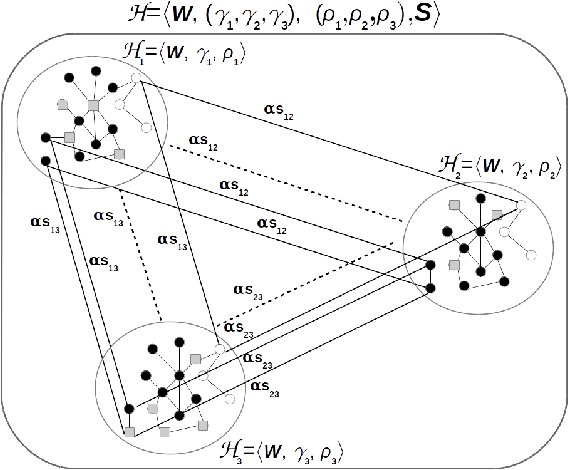

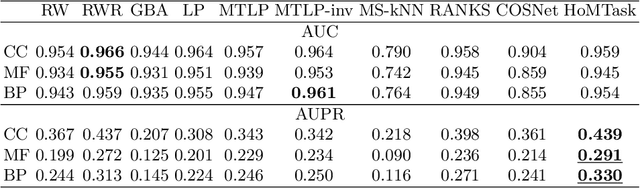
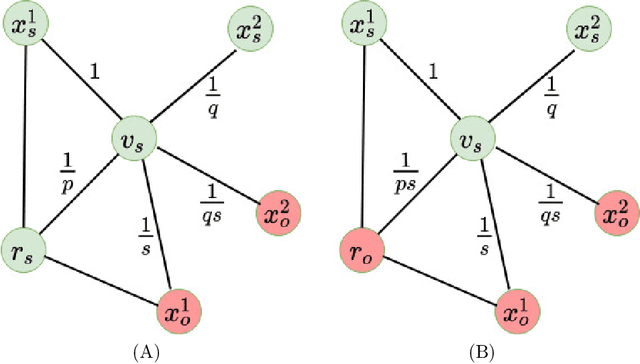
Multitask algorithms typically use task similarity information as a bias to speed up and improve the performance of learning processes. Tasks are learned jointly, sharing information across them, in order to construct models more accurate than those learned separately over single tasks. In this contribution, we present the first multitask model, to our knowledge, based on Hopfield Networks (HNs), named HoMTask. We show that by appropriately building a unique HN embedding all tasks, a more robust and effective classification model can be learned. HoMTask is a transductive semi-supervised parametric HN, that minimizes an energy function extended to all nodes and to all tasks under study. We provide theoretical evidence that the optimal parameters automatically estimated by HoMTask make coherent the model itself with the prior knowledge (connection weights and node labels). The convergence properties of HNs are preserved, and the fixed point reached by the network dynamics gives rise to the prediction of unlabeled nodes. The proposed model improves the classification abilities of singletask HNs on a preliminary benchmark comparison, and achieves competitive performance with state-of-the-art semi-supervised graph-based algorithms.
Notes on hierarchical ensemble methods for DAG-structured taxonomies
Jun 18, 2014


Several real problems ranging from text classification to computational biology are characterized by hierarchical multi-label classification tasks. Most of the methods presented in literature focused on tree-structured taxonomies, but only few on taxonomies structured according to a Directed Acyclic Graph (DAG). In this contribution novel classification ensemble algorithms for DAG-structured taxonomies are introduced. In particular Hierarchical Top-Down (HTD-DAG) and True Path Rule (TPR-DAG) for DAGs are presented and discussed.