 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgentic Mixture-of-Workflows for Multi-Modal Chemical Search
Feb 26, 2025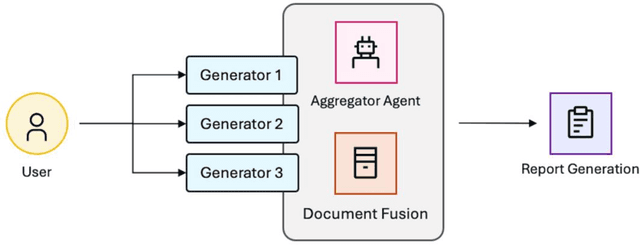
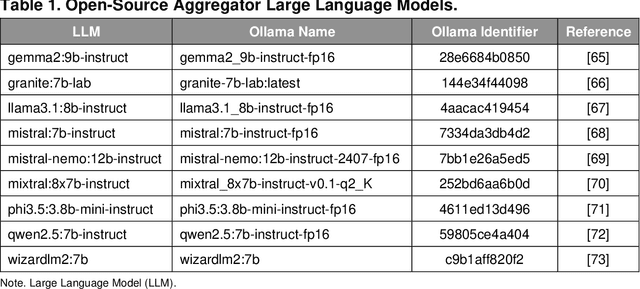
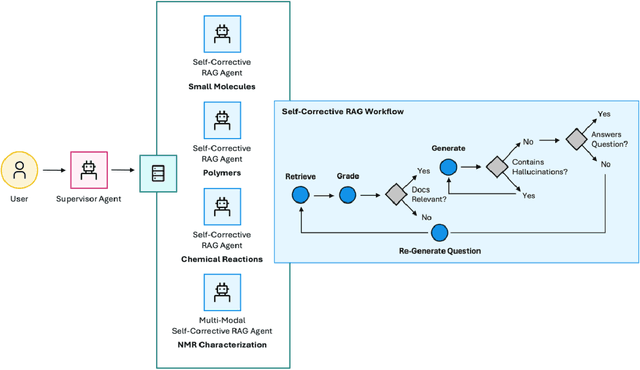
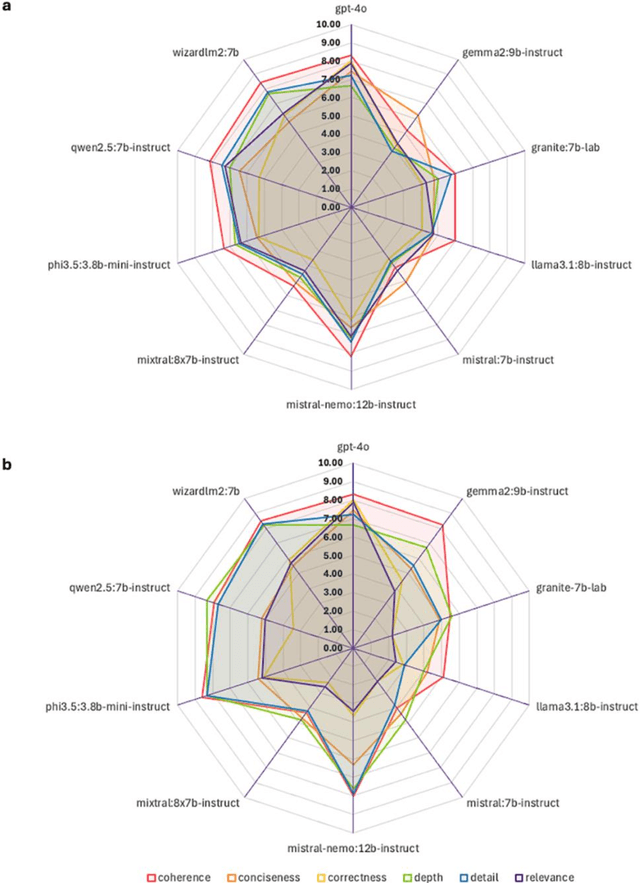
The vast and complex materials design space demands innovative strategies to integrate multidisciplinary scientific knowledge and optimize materials discovery. While large language models (LLMs) have demonstrated promising reasoning and automation capabilities across various domains, their application in materials science remains limited due to a lack of benchmarking standards and practical implementation frameworks. To address these challenges, we introduce Mixture-of-Workflows for Self-Corrective Retrieval-Augmented Generation (CRAG-MoW) - a novel paradigm that orchestrates multiple agentic workflows employing distinct CRAG strategies using open-source LLMs. Unlike prior approaches, CRAG-MoW synthesizes diverse outputs through an orchestration agent, enabling direct evaluation of multiple LLMs across the same problem domain. We benchmark CRAG-MoWs across small molecules, polymers, and chemical reactions, as well as multi-modal nuclear magnetic resonance (NMR) spectral retrieval. Our results demonstrate that CRAG-MoWs achieve performance comparable to GPT-4o while being preferred more frequently in comparative evaluations, highlighting the advantage of structured retrieval and multi-agent synthesis. By revealing performance variations across data types, CRAG-MoW provides a scalable, interpretable, and benchmark-driven approach to optimizing AI architectures for materials discovery. These insights are pivotal in addressing fundamental gaps in benchmarking LLMs and autonomous AI agents for scientific applications.
Leveraging Chemistry Foundation Models to Facilitate Structure Focused Retrieval Augmented Generation in Multi-Agent Workflows for Catalyst and Materials Design
Aug 21, 2024



Molecular property prediction and generative design via deep learning models has been the subject of intense research given its potential to accelerate development of new, high-performance materials. More recently, these workflows have been significantly augmented with the advent of large language models (LLMs) and systems of LLM-driven agents capable of utilizing pre-trained models to make predictions in the context of more complex research tasks. While effective, there is still room for substantial improvement within the agentic systems on the retrieval of salient information for material design tasks. Moreover, alternative uses of predictive deep learning models, such as leveraging their latent representations to facilitate cross-modal retrieval augmented generation within agentic systems to enable task-specific materials design, has remained unexplored. Herein, we demonstrate that large, pre-trained chemistry foundation models can serve as a basis for enabling semantic chemistry information retrieval for both small-molecules, complex polymeric materials, and reactions. Additionally, we show the use of chemistry foundation models in conjunction with image models such as OpenCLIP facilitate unprecedented queries and information retrieval across multiple characterization data domains. Finally, we demonstrate the integration of these systems within multi-agent systems to facilitate structure and topological-based natural language queries and information retrieval for complex research tasks.
RNA-KG: An ontology-based knowledge graph for representing interactions involving RNA molecules
Nov 30, 2023The "RNA world" represents a novel frontier for the study of fundamental biological processes and human diseases and is paving the way for the development of new drugs tailored to the patient's biomolecular characteristics. Although scientific data about coding and non-coding RNA molecules are continuously produced and available from public repositories, they are scattered across different databases and a centralized, uniform, and semantically consistent representation of the "RNA world" is still lacking. We propose RNA-KG, a knowledge graph encompassing biological knowledge about RNAs gathered from more than 50 public databases, integrating functional relationships with genes, proteins, and chemicals and ontologically grounded biomedical concepts. To develop RNA-KG, we first identified, pre-processed, and characterized each data source; next, we built a meta-graph that provides an ontological description of the KG by representing all the bio-molecular entities and medical concepts of interest in this domain, as well as the types of interactions connecting them. Finally, we leveraged an instance-based semantically abstracted knowledge model to specify the ontological alignment according to which RNA-KG was generated. RNA-KG can be downloaded in different formats and also queried by a SPARQL endpoint. A thorough topological analysis of the resulting heterogeneous graph provides further insights into the characteristics of the "RNA world". RNA-KG can be both directly explored and visualized, and/or analyzed by applying computational methods to infer bio-medical knowledge from its heterogeneous nodes and edges. The resource can be easily updated with new experimental data, and specific views of the overall KG can be extracted according to the bio-medical problem to be studied.
An Open-Source Knowledge Graph Ecosystem for the Life Sciences
Jul 11, 2023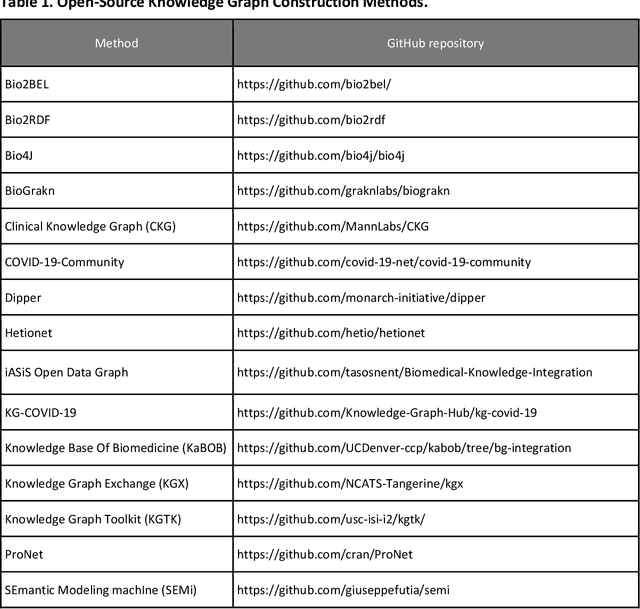
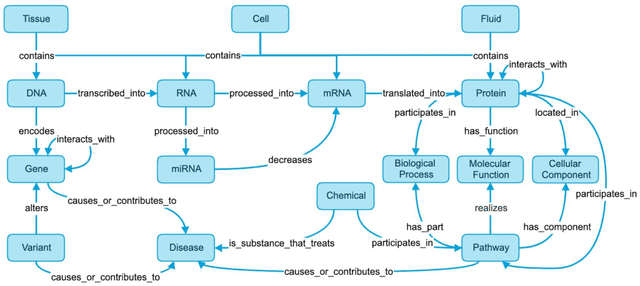
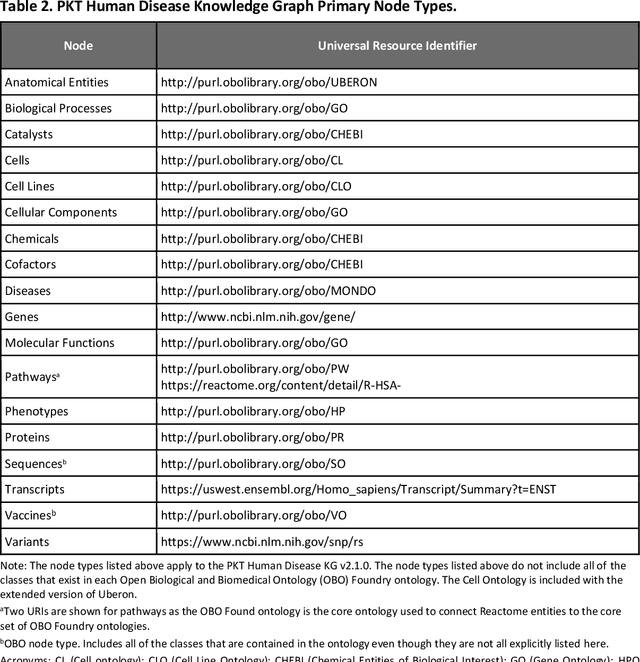
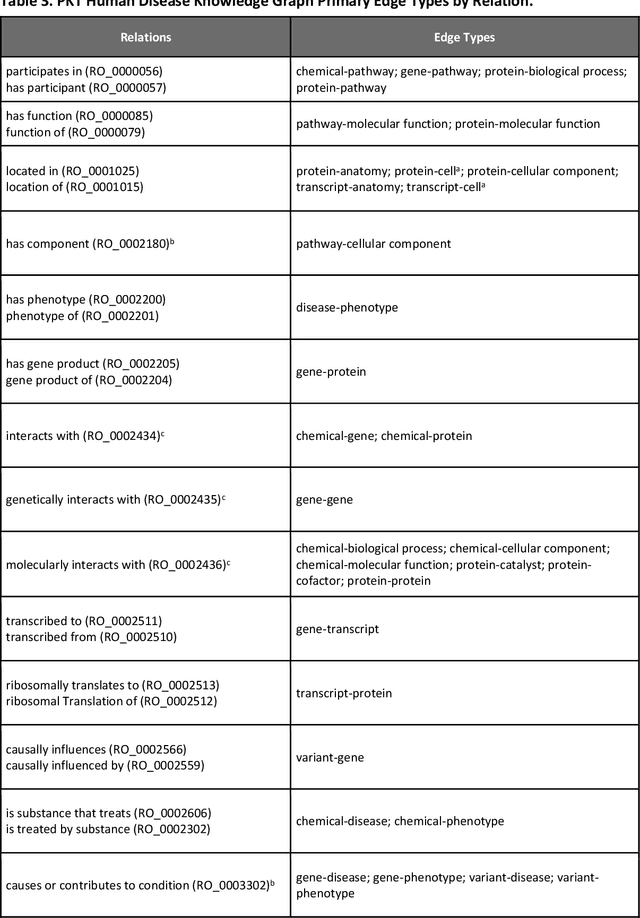
Translational research requires data at multiple scales of biological organization. Advancements in sequencing and multi-omics technologies have increased the availability of these data but researchers face significant integration challenges. Knowledge graphs (KGs) are used to model complex phenomena, and methods exist to automatically construct them. However, tackling complex biomedical integration problems requires flexibility in the way knowledge is modeled. Moreover, existing KG construction methods provide robust tooling at the cost of fixed or limited choices among knowledge representation models. PheKnowLator (Phenotype Knowledge Translator) is a semantic ecosystem for automating the FAIR (Findable, Accessible, Interoperable, and Reusable) construction of ontologically grounded KGs with fully customizable knowledge representation. The ecosystem includes KG construction resources (e.g., data preparation APIs), analysis tools (e.g., SPARQL endpoints and abstraction algorithms), and benchmarks (e.g., prebuilt KGs and embeddings). We evaluate the ecosystem by surveying open-source KG construction methods and analyzing its computational performance when constructing 12 large-scale KGs. With flexible knowledge representation, PheKnowLator enables fully customizable KGs without compromising performance or usability.
Developing a Knowledge Graph Framework for Pharmacokinetic Natural Product-Drug Interactions
Sep 24, 2022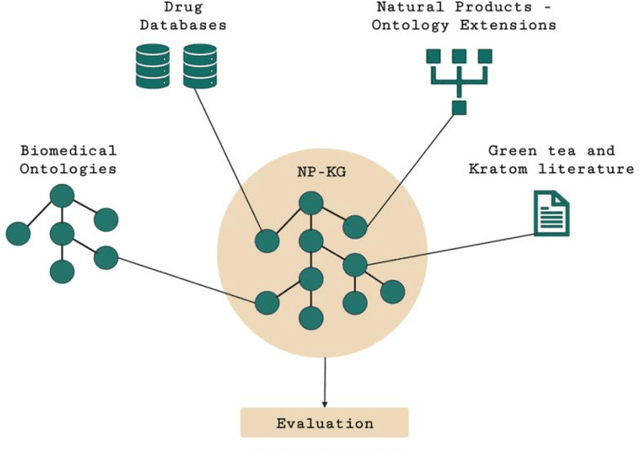
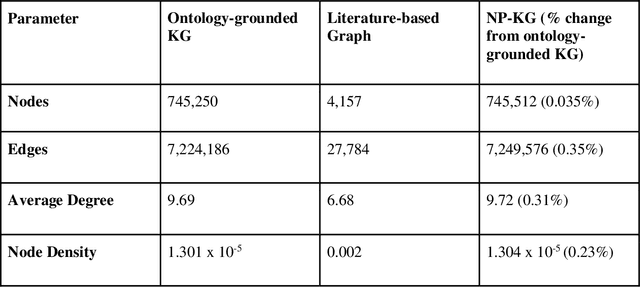
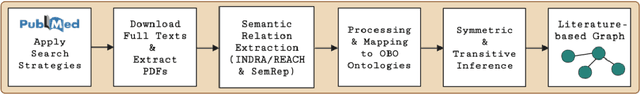
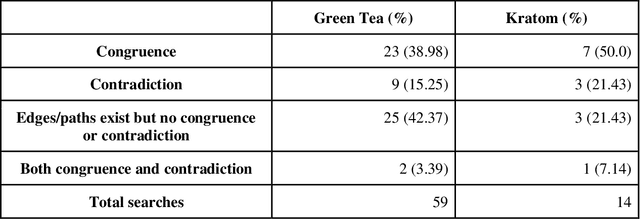
Pharmacokinetic natural product-drug interactions (NPDIs) occur when botanical natural products are co-consumed with pharmaceutical drugs. Understanding mechanisms of NPDIs is key to preventing adverse events. We constructed a knowledge graph framework, NP-KG, as a step toward computational discovery of pharmacokinetic NPDIs. NP-KG is a heterogeneous KG with biomedical ontologies, linked data, and full texts of the scientific literature, constructed with the Phenotype Knowledge Translator framework and the semantic relation extraction systems, SemRep and Integrated Network and Dynamic Reasoning Assembler. NP-KG was evaluated with case studies of pharmacokinetic green tea- and kratom-drug interactions through path searches and meta-path discovery to determine congruent and contradictory information compared to ground truth data. The fully integrated NP-KG consisted of 745,512 nodes and 7,249,576 edges. Evaluation of NP-KG resulted in congruent (38.98% for green tea, 50% for kratom), contradictory (15.25% for green tea, 21.43% for kratom), and both congruent and contradictory (15.25% for green tea, 21.43% for kratom) information. Potential pharmacokinetic mechanisms for several purported NPDIs, including the green tea-raloxifene, green tea-nadolol, kratom-midazolam, kratom-quetiapine, and kratom-venlafaxine interactions were congruent with the published literature. NP-KG is the first KG to integrate biomedical ontologies with full texts of the scientific literature focused on natural products. We demonstrate the application of NP-KG to identify pharmacokinetic interactions involving enzymes, transporters, and pharmaceutical drugs. We envision that NP-KG will facilitate improved human-machine collaboration to guide researchers in future studies of pharmacokinetic NPDIs. The NP-KG framework is publicly available at https://doi.org/10.5281/zenodo.6814507 and https://github.com/sanyabt/np-kg.
Ontologizing Health Systems Data at Scale: Making Translational Discovery a Reality
Sep 10, 2022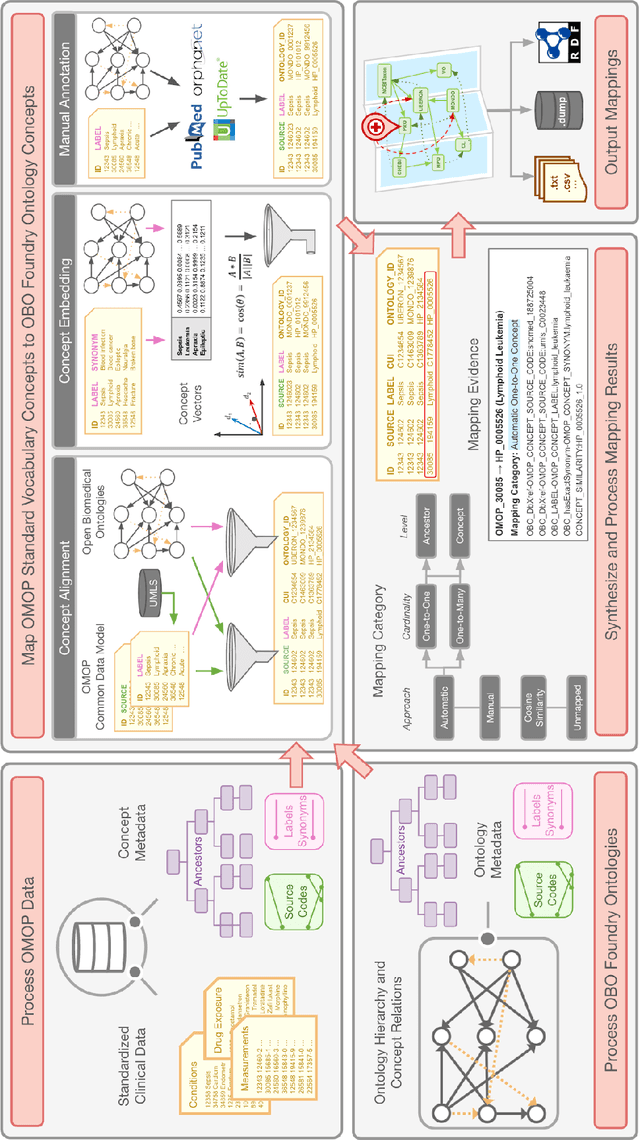
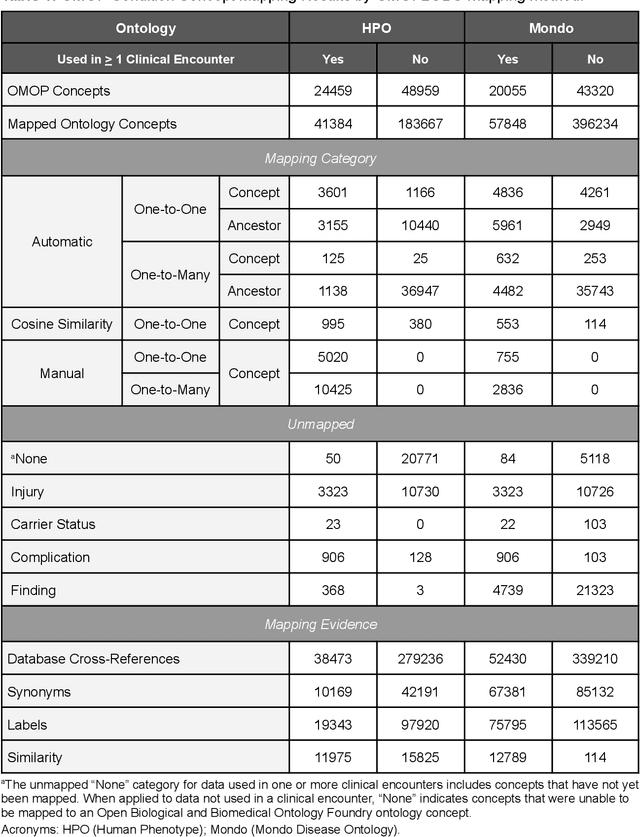
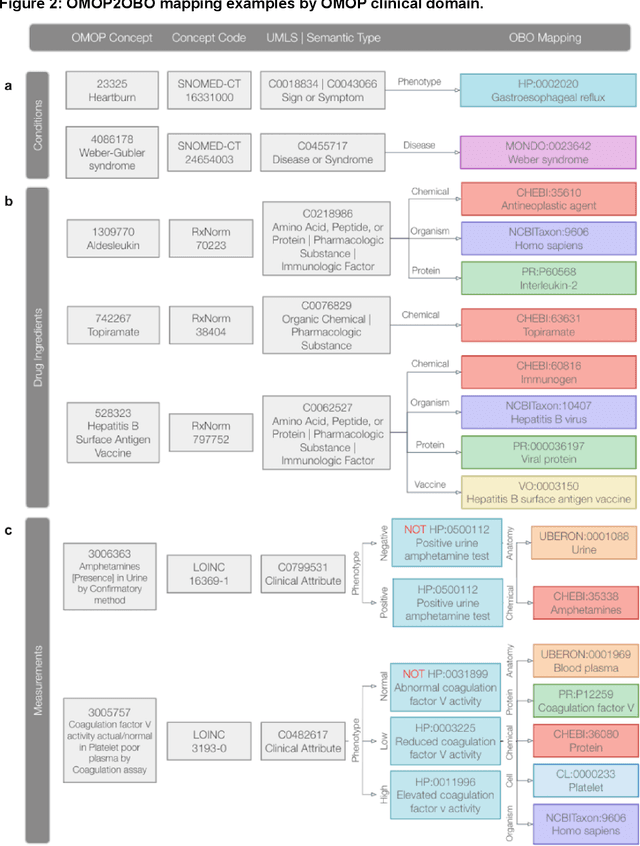
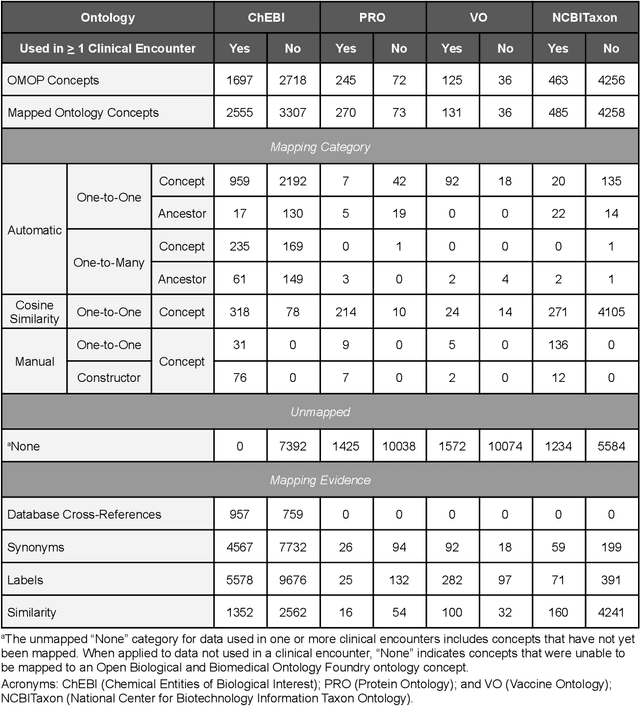
Common data models solve many challenges of standardizing electronic health record (EHR) data, but are unable to semantically integrate the resources needed for deep phenotyping. Open Biological and Biomedical Ontology (OBO) Foundry ontologies provide semantically computable representations of biological knowledge and enable the integration of a variety of biomedical data. However, mapping EHR data to OBO Foundry ontologies requires significant manual curation and domain expertise. We introduce a framework for mapping Observational Medical Outcomes Partnership (OMOP) standard vocabularies to OBO Foundry ontologies. Using this framework, we produced mappings for 92,367 conditions, 8,615 drug ingredients, and 10,673 measurement results. Mapping accuracy was verified by domain experts and when examined across 24 hospitals, the mappings covered 99% of conditions and drug ingredients and 68% of measurements. Finally, we demonstrate that OMOP2OBO mappings can aid in the systematic identification of undiagnosed rare disease patients who might benefit from genetic testing.
Knowledge-Driven Mechanistic Enrichment of the Preeclampsia Ignorome
Jul 28, 2022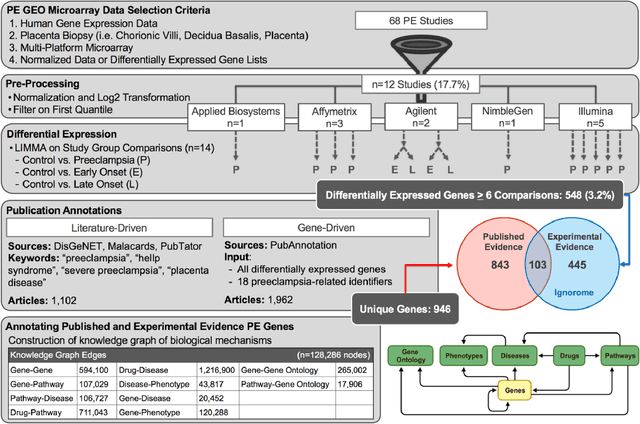
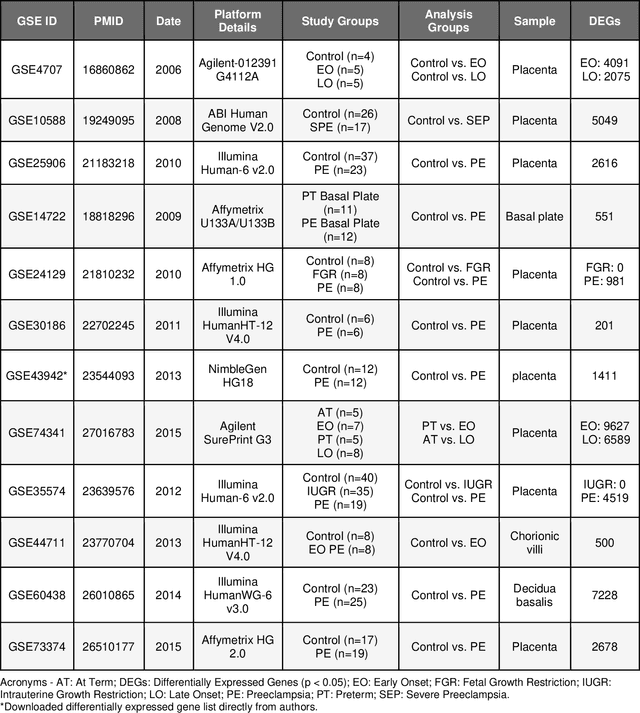
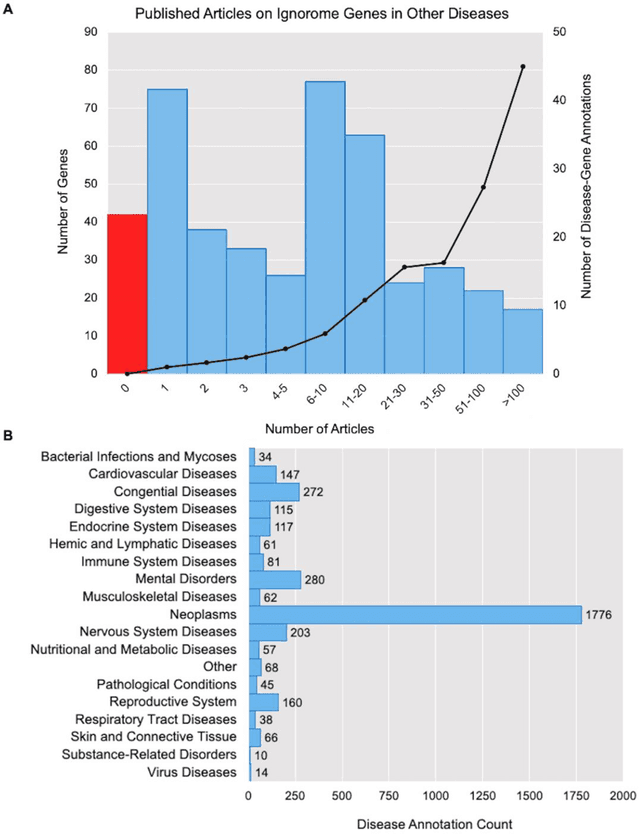
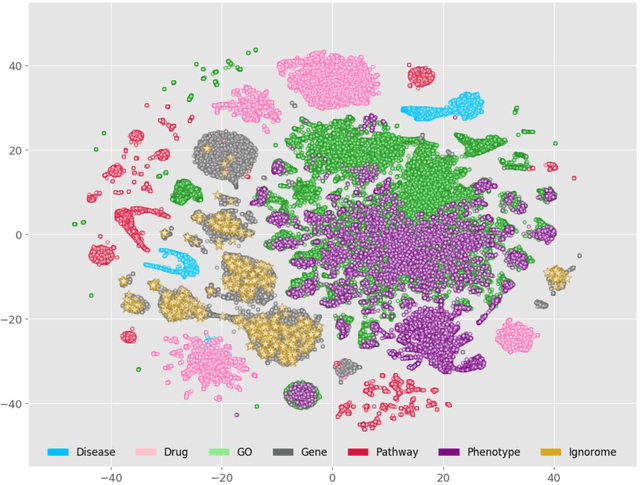
Preeclampsia is a leading cause of maternal and fetal morbidity and mortality. Currently, the only definitive treatment of preeclampsia is delivery of the placenta, which is central to the pathogenesis of the disease. Transcriptional profiling of human placenta from pregnancies complicated by preeclampsia has been extensively performed to identify differentially expressed genes (DEGs). DEGs are identified using unbiased assays, however, the decisions to investigate DEGs experimentally are biased by many factors, causing many DEGs to remain uninvestigated. A set of DEGs which are associated with a disease experimentally, but which have no known association with the disease in the literature is known as the ignorome. Preeclampsia has an extensive body of scientific literature, a large pool of DEG data, and only one definitive treatment. Tools facilitating knowledge-based analyses, which are capable of combining disparate data from many sources in order to suggest underlying mechanisms of action, may be a valuable resource to support discovery and improve our understanding of this disease. In this work we demonstrate how a biomedical knowledge graph (KG) can be used to identify novel preeclampsia molecular mechanisms. Existing open source biomedical resources and publicly available high-throughput transcriptional profiling data were used to identify and annotate the function of currently uninvestigated preeclampsia-associated DEGs. Experimentally investigated genes associated with preeclampsia were identified from PubMed abstracts using text-mining methodologies. The relative complement of the text-mined- and meta-analysis-derived lists were identified as the uninvestigated preeclampsia-associated DEGs (n=445), i.e., the preeclampsia ignorome. Using the KG to investigate relevant DEGs revealed 53 novel clinically relevant and biologically actionable mechanistic associations.
A Methodological Framework for the Comparative Evaluation of Multiple Imputation Methods: Multiple Imputation of Race, Ethnicity and Body Mass Index in the U.S. National COVID Cohort Collaborative
Jun 13, 2022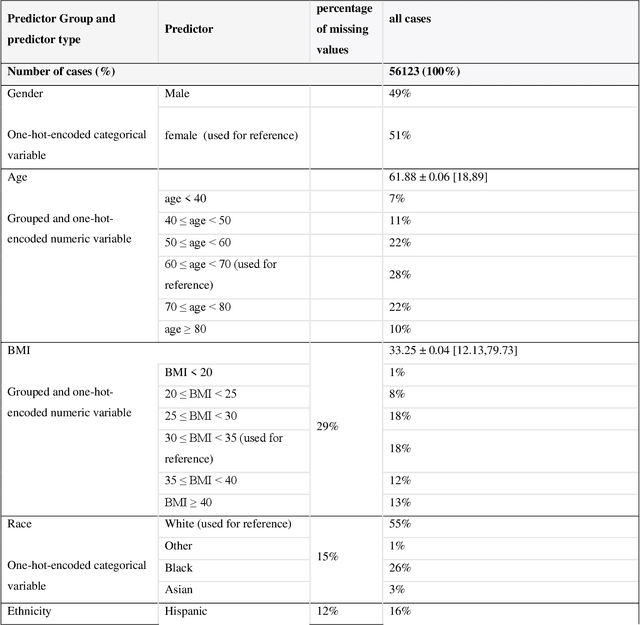
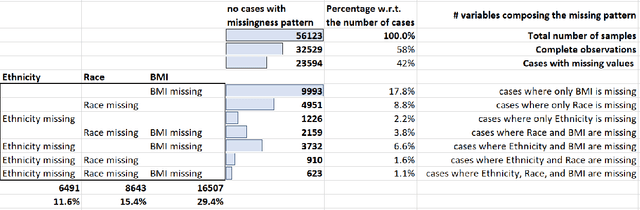

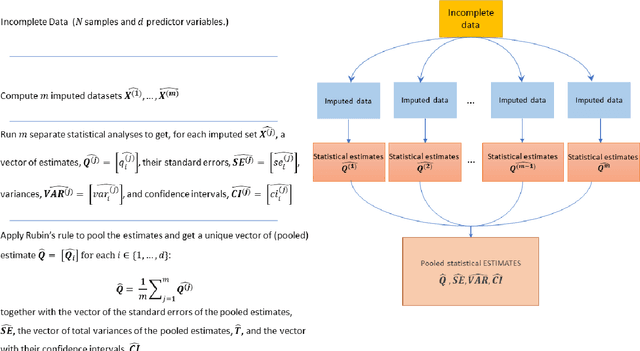
While electronic health records are a rich data source for biomedical research, these systems are not implemented uniformly across healthcare settings and significant data may be missing due to healthcare fragmentation and lack of interoperability between siloed electronic health records. Considering that the deletion of cases with missing data may introduce severe bias in the subsequent analysis, several authors prefer applying a multiple imputation strategy to recover the missing information. Unfortunately, although several literature works have documented promising results by using any of the different multiple imputation algorithms that are now freely available for research, there is no consensus on which MI algorithm works best. Beside the choice of the MI strategy, the choice of the imputation algorithm and its application settings are also both crucial and challenging. In this paper, inspired by the seminal works of Rubin and van Buuren, we propose a methodological framework that may be applied to evaluate and compare several multiple imputation techniques, with the aim to choose the most valid for computing inferences in a clinical research work. Our framework has been applied to validate, and extend on a larger cohort, the results we presented in a previous literature study, where we evaluated the influence of crucial patients' descriptors and COVID-19 severity in patients with type 2 diabetes mellitus whose data is provided by the National COVID Cohort Collaborative Enclave.
GraPE: fast and scalable Graph Processing and Embedding
Oct 12, 2021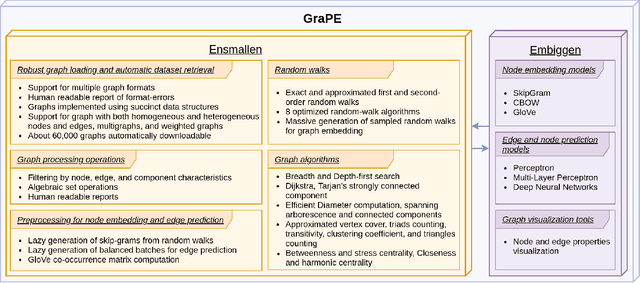
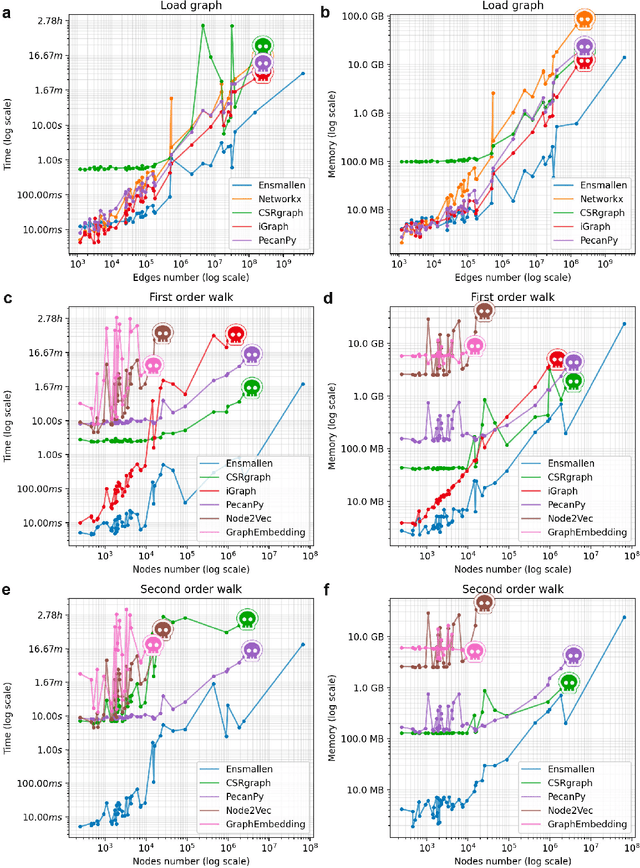
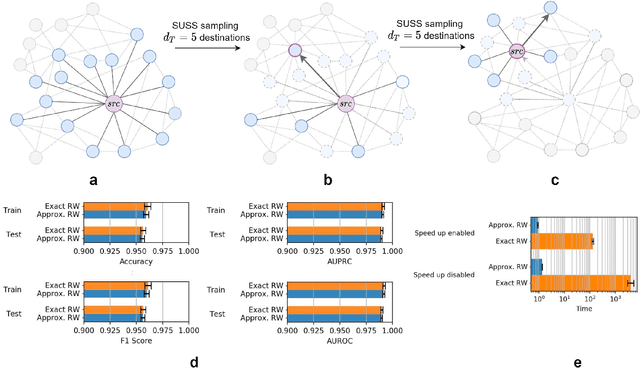
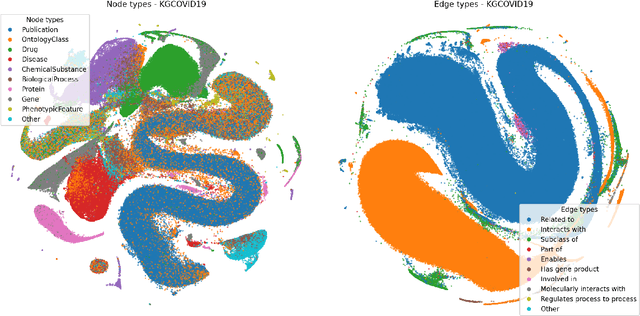
Graph Representation Learning methods have enabled a wide range of learning problems to be addressed for data that can be represented in graph form. Nevertheless, several real world problems in economy, biology, medicine and other fields raised relevant scaling problems with existing methods and their software implementation, due to the size of real world graphs characterized by millions of nodes and billions of edges. We present GraPE, a software resource for graph processing and random walk based embedding, that can scale with large and high-degree graphs and significantly speed up-computation. GraPE comprises specialized data structures, algorithms, and a fast parallel implementation that displays everal orders of magnitude improvement in empirical space and time complexity compared to state of the art software resources, with a corresponding boost in the performance of machine learning methods for edge and node label prediction and for the unsupervised analysis of graphs.GraPE is designed to run on laptop and desktop computers, as well as on high performance computing clusters
A Biomedically oriented automatically annotated Twitter COVID-19 Dataset
Jul 27, 2021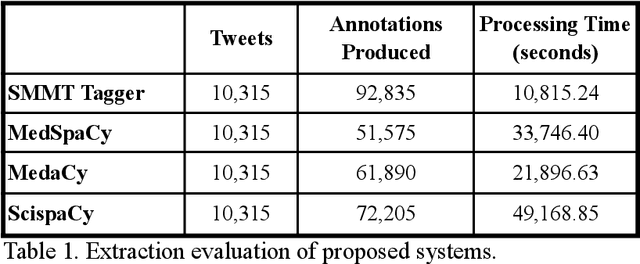
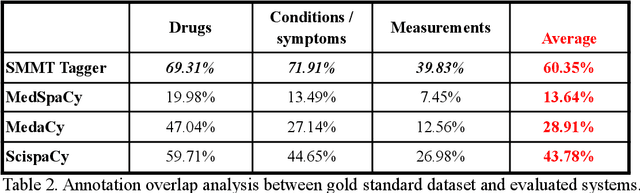
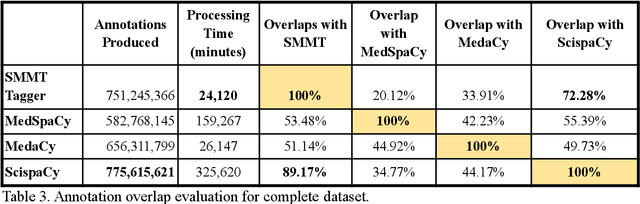
The use of social media data, like Twitter, for biomedical research has been gradually increasing over the years. With the COVID-19 pandemic, researchers have turned to more nontraditional sources of clinical data to characterize the disease in near real-time, study the societal implications of interventions, as well as the sequelae that recovered COVID-19 cases present (Long-COVID). However, manually curated social media datasets are difficult to come by due to the expensive costs of manual annotation and the efforts needed to identify the correct texts. When datasets are available, they are usually very small and their annotations do not generalize well over time or to larger sets of documents. As part of the 2021 Biomedical Linked Annotation Hackathon, we release our dataset of over 120 million automatically annotated tweets for biomedical research purposes. Incorporating best practices, we identify tweets with potentially high clinical relevance. We evaluated our work by comparing several SpaCy-based annotation frameworks against a manually annotated gold-standard dataset. Selecting the best method to use for automatic annotation, we then annotated 120 million tweets and released them publicly for future downstream usage within the biomedical domain.