 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDecomposition Sampling for Efficient Region Annotations in Active Learning
Dec 08, 2025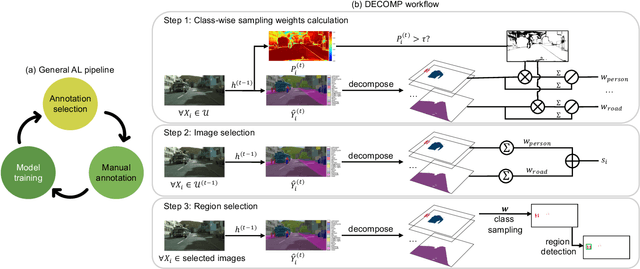
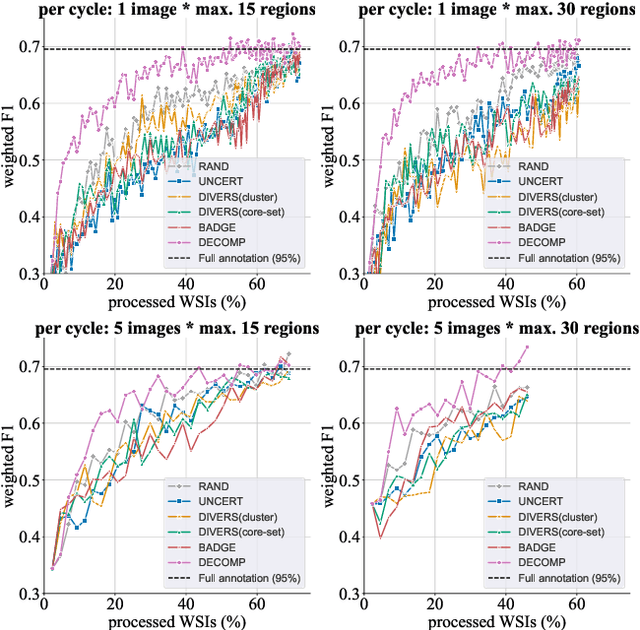
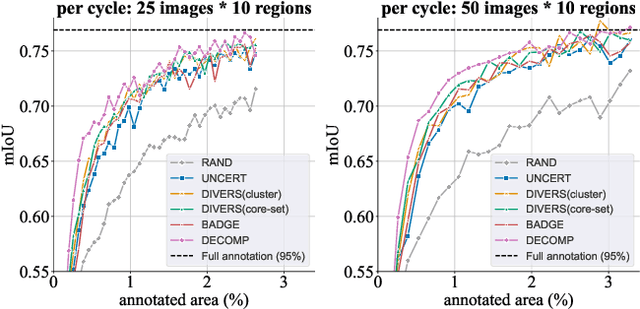
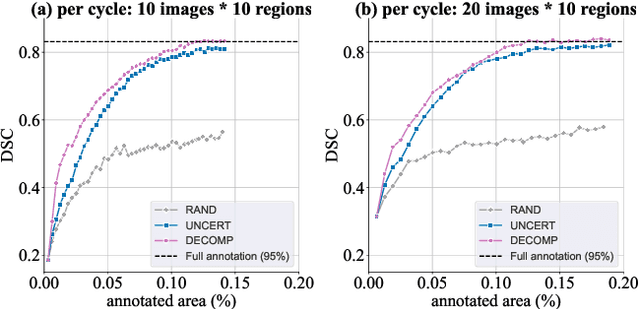
Active learning improves annotation efficiency by selecting the most informative samples for annotation and model training. While most prior work has focused on selecting informative images for classification tasks, we investigate the more challenging setting of dense prediction, where annotations are more costly and time-intensive, especially in medical imaging. Region-level annotation has been shown to be more efficient than image-level annotation for these tasks. However, existing methods for representative annotation region selection suffer from high computational and memory costs, irrelevant region choices, and heavy reliance on uncertainty sampling. We propose decomposition sampling (DECOMP), a new active learning sampling strategy that addresses these limitations. It enhances annotation diversity by decomposing images into class-specific components using pseudo-labels and sampling regions from each class. Class-wise predictive confidence further guides the sampling process, ensuring that difficult classes receive additional annotations. Across ROI classification, 2-D segmentation, and 3-D segmentation, DECOMP consistently surpasses baseline methods by better sampling minority-class regions and boosting performance on these challenging classes. Code is in https://github.com/JingnaQiu/DECOMP.git.
SWAN -- Enabling Fast and Mobile Histopathology Image Annotation through Swipeable Interfaces
Nov 11, 2025The annotation of large scale histopathology image datasets remains a major bottleneck in developing robust deep learning models for clinically relevant tasks, such as mitotic figure classification. Folder-based annotation workflows are usually slow, fatiguing, and difficult to scale. To address these challenges, we introduce SWipeable ANnotations (SWAN), an open-source, MIT-licensed web application that enables intuitive image patch classification using a swiping gesture. SWAN supports both desktop and mobile platforms, offers real-time metadata capture, and allows flexible mapping of swipe gestures to class labels. In a pilot study with four pathologists annotating 600 mitotic figure image patches, we compared SWAN against a traditional folder-sorting workflow. SWAN enabled rapid annotations with pairwise percent agreement ranging from 86.52% to 93.68% (Cohen's Kappa = 0.61-0.80), while for the folder-based method, the pairwise percent agreement ranged from 86.98% to 91.32% (Cohen's Kappa = 0.63-0.75) for the task of classifying atypical versus normal mitotic figures, demonstrating high consistency between annotators and comparable performance. Participants rated the tool as highly usable and appreciated the ability to annotate on mobile devices. These results suggest that SWAN can accelerate image annotation while maintaining annotation quality, offering a scalable and user-friendly alternative to conventional workflows.
Faster, Self-Supervised Super-Resolution for Anisotropic Multi-View MRI Using a Sparse Coordinate Loss
Sep 09, 2025Acquiring images in high resolution is often a challenging task. Especially in the medical sector, image quality has to be balanced with acquisition time and patient comfort. To strike a compromise between scan time and quality for Magnetic Resonance (MR) imaging, two anisotropic scans with different low-resolution (LR) orientations can be acquired. Typically, LR scans are analyzed individually by radiologists, which is time consuming and can lead to inaccurate interpretation. To tackle this, we propose a novel approach for fusing two orthogonal anisotropic LR MR images to reconstruct anatomical details in a unified representation. Our multi-view neural network is trained in a self-supervised manner, without requiring corresponding high-resolution (HR) data. To optimize the model, we introduce a sparse coordinate-based loss, enabling the integration of LR images with arbitrary scaling. We evaluate our method on MR images from two independent cohorts. Our results demonstrate comparable or even improved super-resolution (SR) performance compared to state-of-the-art (SOTA) self-supervised SR methods for different upsampling scales. By combining a patient-agnostic offline and a patient-specific online phase, we achieve a substantial speed-up of up to ten times for patient-specific reconstruction while achieving similar or better SR quality. Code is available at https://github.com/MajaSchle/tripleSR.
Effortless Vision-Language Model Specialization in Histopathology without Annotation
Aug 11, 2025Recent advances in Vision-Language Models (VLMs) in histopathology, such as CONCH and QuiltNet, have demonstrated impressive zero-shot classification capabilities across various tasks. However, their general-purpose design may lead to suboptimal performance in specific downstream applications. While supervised fine-tuning methods address this issue, they require manually labeled samples for adaptation. This paper investigates annotation-free adaptation of VLMs through continued pretraining on domain- and task-relevant image-caption pairs extracted from existing databases. Our experiments on two VLMs, CONCH and QuiltNet, across three downstream tasks reveal that these pairs substantially enhance both zero-shot and few-shot performance. Notably, with larger training sizes, continued pretraining matches the performance of few-shot methods while eliminating manual labeling. Its effectiveness, task-agnostic design, and annotation-free workflow make it a promising pathway for adapting VLMs to new histopathology tasks. Code is available at https://github.com/DeepMicroscopy/Annotation-free-VLM-specialization.
Benchmarking Deep Learning and Vision Foundation Models for Atypical vs. Normal Mitosis Classification with Cross-Dataset Evaluation
Jun 26, 2025Atypical mitoses mark a deviation in the cell division process that can be an independent prognostically relevant marker for tumor malignancy. However, their identification remains challenging due to low prevalence, at times subtle morphological differences from normal mitoses, low inter-rater agreement among pathologists, and class imbalance in datasets. Building on the Atypical Mitosis dataset for Breast Cancer (AMi-Br), this study presents a comprehensive benchmark comparing deep learning approaches for automated atypical mitotic figure (AMF) classification, including baseline models, foundation models with linear probing, and foundation models fine-tuned with low-rank adaptation (LoRA). For rigorous evaluation, we further introduce two new hold-out AMF datasets - AtNorM-Br, a dataset of mitoses from the The TCGA breast cancer cohort, and AtNorM-MD, a multi-domain dataset of mitoses from the MIDOG++ training set. We found average balanced accuracy values of up to 0.8135, 0.7696, and 0.7705 on the in-domain AMi-Br and the out-of-domain AtNorm-Br and AtNorM-MD datasets, respectively, with the results being particularly good for LoRA-based adaptation of the Virchow-line of foundation models. Our work shows that atypical mitosis classification, while being a challenging problem, can be effectively addressed through the use of recent advances in transfer learning and model fine-tuning techniques. We make available all code and data used in this paper in this github repository: https://github.com/DeepMicroscopy/AMi-Br_Benchmark.
Dataset Distillation with Probabilistic Latent Features
May 10, 2025As deep learning models grow in complexity and the volume of training data increases, reducing storage and computational costs becomes increasingly important. Dataset distillation addresses this challenge by synthesizing a compact set of synthetic data that can effectively replace the original dataset in downstream classification tasks. While existing methods typically rely on mapping data from pixel space to the latent space of a generative model, we propose a novel stochastic approach that models the joint distribution of latent features. This allows our method to better capture spatial structures and produce diverse synthetic samples, which benefits model training. Specifically, we introduce a low-rank multivariate normal distribution parameterized by a lightweight network. This design maintains low computational complexity and is compatible with various matching networks used in dataset distillation. After distillation, synthetic images are generated by feeding the learned latent features into a pretrained generator. These synthetic images are then used to train classification models, and performance is evaluated on real test set. We validate our method on several benchmarks, including ImageNet subsets, CIFAR-10, and the MedMNIST histopathological dataset. Our approach achieves state-of-the-art cross architecture performance across a range of backbone architectures, demonstrating its generality and effectiveness.
CyclePose -- Leveraging Cycle-Consistency for Annotation-Free Nuclei Segmentation in Fluorescence Microscopy
Mar 14, 2025In recent years, numerous neural network architectures specifically designed for the instance segmentation of nuclei in microscopic images have been released. These models embed nuclei-specific priors to outperform generic architectures like U-Nets; however, they require large annotated datasets, which are often not available. Generative models (GANs, diffusion models) have been used to compensate for this by synthesizing training data. These two-stage approaches are computationally expensive, as first a generative model and then a segmentation model has to be trained. We propose CyclePose, a hybrid framework integrating synthetic data generation and segmentation training. CyclePose builds on a CycleGAN architecture, which allows unpaired translation between microscopy images and segmentation masks. We embed a segmentation model into CycleGAN and leverage a cycle consistency loss for self-supervision. Without annotated data, CyclePose outperforms other weakly or unsupervised methods on two public datasets. Code is available at https://github.com/jonasutz/CyclePose
DiffRenderGAN: Addressing Training Data Scarcity in Deep Segmentation Networks for Quantitative Nanomaterial Analysis through Differentiable Rendering and Generative Modelling
Feb 13, 2025Nanomaterials exhibit distinctive properties governed by parameters such as size, shape, and surface characteristics, which critically influence their applications and interactions across technological, biological, and environmental contexts. Accurate quantification and understanding of these materials are essential for advancing research and innovation. In this regard, deep learning segmentation networks have emerged as powerful tools that enable automated insights and replace subjective methods with precise quantitative analysis. However, their efficacy depends on representative annotated datasets, which are challenging to obtain due to the costly imaging of nanoparticles and the labor-intensive nature of manual annotations. To overcome these limitations, we introduce DiffRenderGAN, a novel generative model designed to produce annotated synthetic data. By integrating a differentiable renderer into a Generative Adversarial Network (GAN) framework, DiffRenderGAN optimizes textural rendering parameters to generate realistic, annotated nanoparticle images from non-annotated real microscopy images. This approach reduces the need for manual intervention and enhances segmentation performance compared to existing synthetic data methods by generating diverse and realistic data. Tested on multiple ion and electron microscopy cases, including titanium dioxide (TiO$_2$), silicon dioxide (SiO$_2$)), and silver nanowires (AgNW), DiffRenderGAN bridges the gap between synthetic and real data, advancing the quantification and understanding of complex nanomaterial systems.
A Self-supervised Multimodal Deep Learning Approach to Differentiate Post-radiotherapy Progression from Pseudoprogression in Glioblastoma
Feb 06, 2025



Accurate differentiation of pseudoprogression (PsP) from True Progression (TP) following radiotherapy (RT) in glioblastoma (GBM) patients is crucial for optimal treatment planning. However, this task remains challenging due to the overlapping imaging characteristics of PsP and TP. This study therefore proposes a multimodal deep-learning approach utilizing complementary information from routine anatomical MR images, clinical parameters, and RT treatment planning information for improved predictive accuracy. The approach utilizes a self-supervised Vision Transformer (ViT) to encode multi-sequence MR brain volumes to effectively capture both global and local context from the high dimensional input. The encoder is trained in a self-supervised upstream task on unlabeled glioma MRI datasets from the open BraTS2021, UPenn-GBM, and UCSF-PDGM datasets to generate compact, clinically relevant representations from FLAIR and T1 post-contrast sequences. These encoded MR inputs are then integrated with clinical data and RT treatment planning information through guided cross-modal attention, improving progression classification accuracy. This work was developed using two datasets from different centers: the Burdenko Glioblastoma Progression Dataset (n = 59) for training and validation, and the GlioCMV progression dataset from the University Hospital Erlangen (UKER) (n = 20) for testing. The proposed method achieved an AUC of 75.3%, outperforming the current state-of-the-art data-driven approaches. Importantly, the proposed approach relies on readily available anatomical MRI sequences, clinical data, and RT treatment planning information, enhancing its clinical feasibility. The proposed approach addresses the challenge of limited data availability for PsP and TP differentiation and could allow for improved clinical decision-making and optimized treatment plans for GBM patients.
Is Self-Supervision Enough? Benchmarking Foundation Models Against End-to-End Training for Mitotic Figure Classification
Dec 09, 2024


Foundation models (FMs), i.e., models trained on a vast amount of typically unlabeled data, have become popular and available recently for the domain of histopathology. The key idea is to extract semantically rich vectors from any input patch, allowing for the use of simple subsequent classification networks potentially reducing the required amounts of labeled data, and increasing domain robustness. In this work, we investigate to which degree this also holds for mitotic figure classification. Utilizing two popular public mitotic figure datasets, we compared linear probing of five publicly available FMs against models trained on ImageNet and a simple ResNet50 end-to-end-trained baseline. We found that the end-to-end-trained baseline outperformed all FM-based classifiers, regardless of the amount of data provided. Additionally, we did not observe the FM-based classifiers to be more robust against domain shifts, rendering both of the above assumptions incorrect.