 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDecomposition Sampling for Efficient Region Annotations in Active Learning
Dec 08, 2025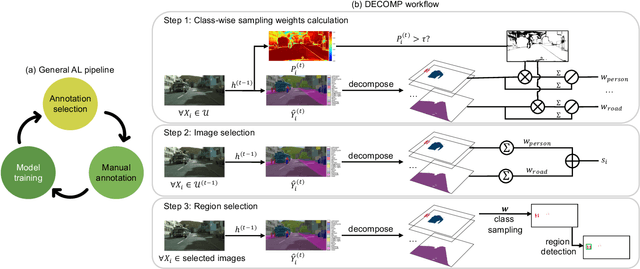
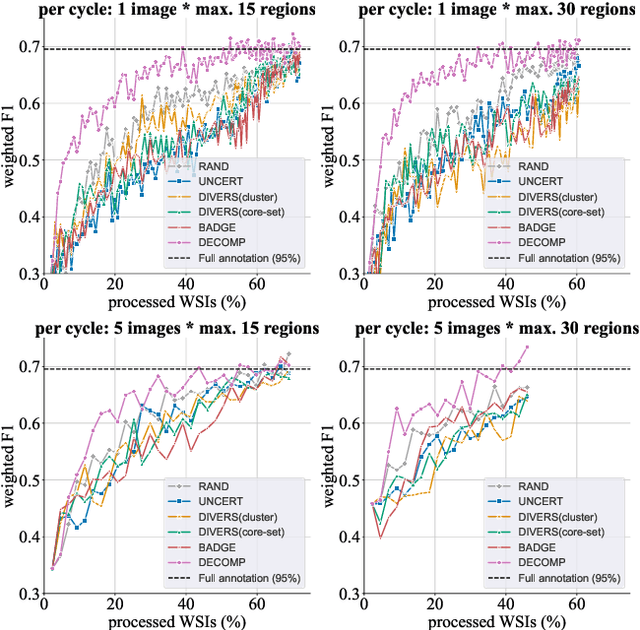
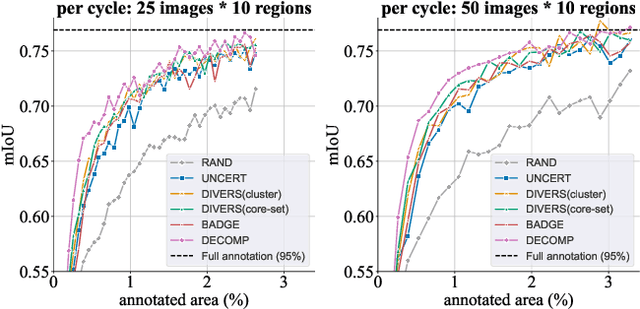
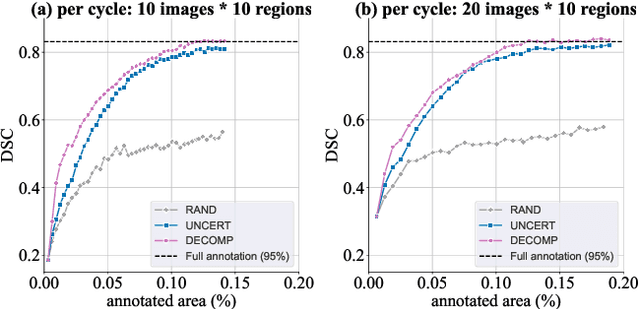
Active learning improves annotation efficiency by selecting the most informative samples for annotation and model training. While most prior work has focused on selecting informative images for classification tasks, we investigate the more challenging setting of dense prediction, where annotations are more costly and time-intensive, especially in medical imaging. Region-level annotation has been shown to be more efficient than image-level annotation for these tasks. However, existing methods for representative annotation region selection suffer from high computational and memory costs, irrelevant region choices, and heavy reliance on uncertainty sampling. We propose decomposition sampling (DECOMP), a new active learning sampling strategy that addresses these limitations. It enhances annotation diversity by decomposing images into class-specific components using pseudo-labels and sampling regions from each class. Class-wise predictive confidence further guides the sampling process, ensuring that difficult classes receive additional annotations. Across ROI classification, 2-D segmentation, and 3-D segmentation, DECOMP consistently surpasses baseline methods by better sampling minority-class regions and boosting performance on these challenging classes. Code is in https://github.com/JingnaQiu/DECOMP.git.
SWAN -- Enabling Fast and Mobile Histopathology Image Annotation through Swipeable Interfaces
Nov 11, 2025The annotation of large scale histopathology image datasets remains a major bottleneck in developing robust deep learning models for clinically relevant tasks, such as mitotic figure classification. Folder-based annotation workflows are usually slow, fatiguing, and difficult to scale. To address these challenges, we introduce SWipeable ANnotations (SWAN), an open-source, MIT-licensed web application that enables intuitive image patch classification using a swiping gesture. SWAN supports both desktop and mobile platforms, offers real-time metadata capture, and allows flexible mapping of swipe gestures to class labels. In a pilot study with four pathologists annotating 600 mitotic figure image patches, we compared SWAN against a traditional folder-sorting workflow. SWAN enabled rapid annotations with pairwise percent agreement ranging from 86.52% to 93.68% (Cohen's Kappa = 0.61-0.80), while for the folder-based method, the pairwise percent agreement ranged from 86.98% to 91.32% (Cohen's Kappa = 0.63-0.75) for the task of classifying atypical versus normal mitotic figures, demonstrating high consistency between annotators and comparable performance. Participants rated the tool as highly usable and appreciated the ability to annotate on mobile devices. These results suggest that SWAN can accelerate image annotation while maintaining annotation quality, offering a scalable and user-friendly alternative to conventional workflows.
Effortless Vision-Language Model Specialization in Histopathology without Annotation
Aug 11, 2025Recent advances in Vision-Language Models (VLMs) in histopathology, such as CONCH and QuiltNet, have demonstrated impressive zero-shot classification capabilities across various tasks. However, their general-purpose design may lead to suboptimal performance in specific downstream applications. While supervised fine-tuning methods address this issue, they require manually labeled samples for adaptation. This paper investigates annotation-free adaptation of VLMs through continued pretraining on domain- and task-relevant image-caption pairs extracted from existing databases. Our experiments on two VLMs, CONCH and QuiltNet, across three downstream tasks reveal that these pairs substantially enhance both zero-shot and few-shot performance. Notably, with larger training sizes, continued pretraining matches the performance of few-shot methods while eliminating manual labeling. Its effectiveness, task-agnostic design, and annotation-free workflow make it a promising pathway for adapting VLMs to new histopathology tasks. Code is available at https://github.com/DeepMicroscopy/Annotation-free-VLM-specialization.
Benchmarking Deep Learning and Vision Foundation Models for Atypical vs. Normal Mitosis Classification with Cross-Dataset Evaluation
Jun 26, 2025Atypical mitoses mark a deviation in the cell division process that can be an independent prognostically relevant marker for tumor malignancy. However, their identification remains challenging due to low prevalence, at times subtle morphological differences from normal mitoses, low inter-rater agreement among pathologists, and class imbalance in datasets. Building on the Atypical Mitosis dataset for Breast Cancer (AMi-Br), this study presents a comprehensive benchmark comparing deep learning approaches for automated atypical mitotic figure (AMF) classification, including baseline models, foundation models with linear probing, and foundation models fine-tuned with low-rank adaptation (LoRA). For rigorous evaluation, we further introduce two new hold-out AMF datasets - AtNorM-Br, a dataset of mitoses from the The TCGA breast cancer cohort, and AtNorM-MD, a multi-domain dataset of mitoses from the MIDOG++ training set. We found average balanced accuracy values of up to 0.8135, 0.7696, and 0.7705 on the in-domain AMi-Br and the out-of-domain AtNorm-Br and AtNorM-MD datasets, respectively, with the results being particularly good for LoRA-based adaptation of the Virchow-line of foundation models. Our work shows that atypical mitosis classification, while being a challenging problem, can be effectively addressed through the use of recent advances in transfer learning and model fine-tuning techniques. We make available all code and data used in this paper in this github repository: https://github.com/DeepMicroscopy/AMi-Br_Benchmark.
A Histologic Dataset of Normal and Atypical Mitotic Figures on Human Breast Cancer (AMi-Br)
Jan 08, 2025Assessment of the density of mitotic figures (MFs) in histologic tumor sections is an important prognostic marker for many tumor types, including breast cancer. Recently, it has been reported in multiple works that the quantity of MFs with an atypical morphology (atypical MFs, AMFs) might be an independent prognostic criterion for breast cancer. AMFs are an indicator of mutations in the genes regulating the cell cycle and can lead to aberrant chromosome constitution (aneuploidy) of the tumor cells. To facilitate further research on this topic using pattern recognition, we present the first ever publicly available dataset of atypical and normal MFs (AMi-Br). For this, we utilized two of the most popular MF datasets (MIDOG 2021 and TUPAC) and subclassified all MFs using a three expert majority vote. Our final dataset consists of 3,720 MFs, split into 832 AMFs (22.4%) and 2,888 normal MFs (77.6%) across all 223 tumor cases in the combined set. We provide baseline classification experiments to investigate the consistency of the dataset, using a Monte Carlo cross-validation and different strategies to combat class imbalance. We found an averaged balanced accuracy of up to 0.806 when using a patch-level data set split, and up to 0.713 when using a patient-level split.
Is Self-Supervision Enough? Benchmarking Foundation Models Against End-to-End Training for Mitotic Figure Classification
Dec 09, 2024


Foundation models (FMs), i.e., models trained on a vast amount of typically unlabeled data, have become popular and available recently for the domain of histopathology. The key idea is to extract semantically rich vectors from any input patch, allowing for the use of simple subsequent classification networks potentially reducing the required amounts of labeled data, and increasing domain robustness. In this work, we investigate to which degree this also holds for mitotic figure classification. Utilizing two popular public mitotic figure datasets, we compared linear probing of five publicly available FMs against models trained on ImageNet and a simple ResNet50 end-to-end-trained baseline. We found that the end-to-end-trained baseline outperformed all FM-based classifiers, regardless of the amount of data provided. Additionally, we did not observe the FM-based classifiers to be more robust against domain shifts, rendering both of the above assumptions incorrect.
Domain and Content Adaptive Convolutions for Cross-Domain Adenocarcinoma Segmentation
Sep 15, 2024Recent advances in computer-aided diagnosis for histopathology have been largely driven by the use of deep learning models for automated image analysis. While these networks can perform on par with medical experts, their performance can be impeded by out-of-distribution data. The Cross-Organ and Cross-Scanner Adenocarcinoma Segmentation (COSAS) challenge aimed to address the task of cross-domain adenocarcinoma segmentation in the presence of morphological and scanner-induced domain shifts. In this paper, we present a U-Net-based segmentation framework designed to tackle this challenge. Our approach achieved segmentation scores of 0.8020 for the cross-organ track and 0.8527 for the cross-scanner track on the final challenge test sets, ranking it the best-performing submission.
Leveraging image captions for selective whole slide image annotation
Jul 08, 2024


Acquiring annotations for whole slide images (WSIs)-based deep learning tasks, such as creating tissue segmentation masks or detecting mitotic figures, is a laborious process due to the extensive image size and the significant manual work involved in the annotation. This paper focuses on identifying and annotating specific image regions that optimize model training, given a limited annotation budget. While random sampling helps capture data variance by collecting annotation regions throughout the WSIs, insufficient data curation may result in an inadequate representation of minority classes. Recent studies proposed diversity sampling to select a set of regions that maximally represent unique characteristics of the WSIs. This is done by pretraining on unlabeled data through self-supervised learning and then clustering all regions in the latent space. However, establishing the optimal number of clusters can be difficult and not all clusters are task-relevant. This paper presents prototype sampling, a new method for annotation region selection. It discovers regions exhibiting typical characteristics of each task-specific class. The process entails recognizing class prototypes from extensive histopathology image-caption databases and detecting unlabeled image regions that resemble these prototypes. Our results show that prototype sampling is more effective than random and diversity sampling in identifying annotation regions with valuable training information, resulting in improved model performance in semantic segmentation and mitotic figure detection tasks. Code is available at https://github.com/DeepMicroscopy/Prototype-sampling.
On the Value of PHH3 for Mitotic Figure Detection on H&E-stained Images
Jun 28, 2024The count of mitotic figures (MFs) observed in hematoxylin and eosin (H&E)-stained slides is an important prognostic marker as it is a measure for tumor cell proliferation. However, the identification of MFs has a known low inter-rater agreement. Deep learning algorithms can standardize this task, but they require large amounts of annotated data for training and validation. Furthermore, label noise introduced during the annotation process may impede the algorithm's performance. Unlike H&E, the mitosis-specific antibody phospho-histone H3 (PHH3) specifically highlights MFs. Counting MFs on slides stained against PHH3 leads to higher agreement among raters and has therefore recently been used as a ground truth for the annotation of MFs in H&E. However, as PHH3 facilitates the recognition of cells indistinguishable from H&E stain alone, the use of this ground truth could potentially introduce noise into the H&E-related dataset, impacting model performance. This study analyzes the impact of PHH3-assisted MF annotation on inter-rater reliability and object level agreement through an extensive multi-rater experiment. We found that the annotators' object-level agreement increased when using PHH3-assisted labeling. Subsequently, MF detectors were evaluated on the resulting datasets to investigate the influence of PHH3-assisted labeling on the models' performance. Additionally, a novel dual-stain MF detector was developed to investigate the interpretation-shift of PHH3-assisted labels used in H&E, which clearly outperformed single-stain detectors. However, the PHH3-assisted labels did not have a positive effect on solely H&E-based models. The high performance of our dual-input detector reveals an information mismatch between the H&E and PHH3-stained images as the cause of this effect.
Model-based Cleaning of the QUILT-1M Pathology Dataset for Text-Conditional Image Synthesis
Apr 11, 2024The QUILT-1M dataset is the first openly available dataset containing images harvested from various online sources. While it provides a huge data variety, the image quality and composition is highly heterogeneous, impacting its utility for text-conditional image synthesis. We propose an automatic pipeline that provides predictions of the most common impurities within the images, e.g., visibility of narrators, desktop environment and pathology software, or text within the image. Additionally, we propose to use semantic alignment filtering of the image-text pairs. Our findings demonstrate that by rigorously filtering the dataset, there is a substantial enhancement of image fidelity in text-to-image tasks.