 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMitosis Detection in the Wild: Multi-Tumor and Context-Aware Generalization in the MIDOG 2025 Challenge
Jun 05, 2026Automated mitosis detection is a well-established task in computational pathology. While previous benchmarks focused on scanner-induced domain shift, clinical "real-world" application requires models to be robust across the vast variance to be expected in the histological landscape. The MItosis DOmain Generalization (MIDOG) 2025 challenge was designed to evaluate algorithmic performance across unprecedented biological and contextual diversity. We curated a test dataset of 365 cases, encompassing 12 distinct human, canine and feline tumor types, digitized across multiple scanning platforms. Moving beyond hand-selected hotspots, the challenge required detection also in random tissue areas (representative of the whole slide detection situation) and challenging areas (areas rich in hard negatives). In the second track, we introduced the classification of atypical mitotic figures (AMFs). There were 18 teams submitting to the detection track, with F1 scores ranging up to 0.740. In the AMF detection track, we had 21 submissions with balanced accuracy values up to 0.908. Our analysis reveals that while most models perform reliably in traditional hotspots, significant performance degradation occurs in challenging ROIs, where false positive rates tripled. Furthermore, performance varied significantly across the 12 tumor types, highlighting "blind spots" in current state-of-the-art architectures when encountering rare or highly pleomorphic malignancies. Moreover, we evaluated the effectiveness of ensembling and found a mean increases of 1.5 and 1.3 percentage points in F1 score and balanced accuracy, respectively. In contrast, TTA showed no relevant improvement. MIDOG 2025 demonstrates that "in the wild" mitosis detection remains a significant hurdle. The transition from hotspot-only evaluation to a multi-contextual framework provides a more realistic proxy for clinical reliability.
SWAN -- Enabling Fast and Mobile Histopathology Image Annotation through Swipeable Interfaces
Nov 11, 2025The annotation of large scale histopathology image datasets remains a major bottleneck in developing robust deep learning models for clinically relevant tasks, such as mitotic figure classification. Folder-based annotation workflows are usually slow, fatiguing, and difficult to scale. To address these challenges, we introduce SWipeable ANnotations (SWAN), an open-source, MIT-licensed web application that enables intuitive image patch classification using a swiping gesture. SWAN supports both desktop and mobile platforms, offers real-time metadata capture, and allows flexible mapping of swipe gestures to class labels. In a pilot study with four pathologists annotating 600 mitotic figure image patches, we compared SWAN against a traditional folder-sorting workflow. SWAN enabled rapid annotations with pairwise percent agreement ranging from 86.52% to 93.68% (Cohen's Kappa = 0.61-0.80), while for the folder-based method, the pairwise percent agreement ranged from 86.98% to 91.32% (Cohen's Kappa = 0.63-0.75) for the task of classifying atypical versus normal mitotic figures, demonstrating high consistency between annotators and comparable performance. Participants rated the tool as highly usable and appreciated the ability to annotate on mobile devices. These results suggest that SWAN can accelerate image annotation while maintaining annotation quality, offering a scalable and user-friendly alternative to conventional workflows.
Benchmarking Deep Learning and Vision Foundation Models for Atypical vs. Normal Mitosis Classification with Cross-Dataset Evaluation
Jun 26, 2025Atypical mitoses mark a deviation in the cell division process that can be an independent prognostically relevant marker for tumor malignancy. However, their identification remains challenging due to low prevalence, at times subtle morphological differences from normal mitoses, low inter-rater agreement among pathologists, and class imbalance in datasets. Building on the Atypical Mitosis dataset for Breast Cancer (AMi-Br), this study presents a comprehensive benchmark comparing deep learning approaches for automated atypical mitotic figure (AMF) classification, including baseline models, foundation models with linear probing, and foundation models fine-tuned with low-rank adaptation (LoRA). For rigorous evaluation, we further introduce two new hold-out AMF datasets - AtNorM-Br, a dataset of mitoses from the The TCGA breast cancer cohort, and AtNorM-MD, a multi-domain dataset of mitoses from the MIDOG++ training set. We found average balanced accuracy values of up to 0.8135, 0.7696, and 0.7705 on the in-domain AMi-Br and the out-of-domain AtNorm-Br and AtNorM-MD datasets, respectively, with the results being particularly good for LoRA-based adaptation of the Virchow-line of foundation models. Our work shows that atypical mitosis classification, while being a challenging problem, can be effectively addressed through the use of recent advances in transfer learning and model fine-tuning techniques. We make available all code and data used in this paper in this github repository: https://github.com/DeepMicroscopy/AMi-Br_Benchmark.
A Histologic Dataset of Normal and Atypical Mitotic Figures on Human Breast Cancer (AMi-Br)
Jan 08, 2025Assessment of the density of mitotic figures (MFs) in histologic tumor sections is an important prognostic marker for many tumor types, including breast cancer. Recently, it has been reported in multiple works that the quantity of MFs with an atypical morphology (atypical MFs, AMFs) might be an independent prognostic criterion for breast cancer. AMFs are an indicator of mutations in the genes regulating the cell cycle and can lead to aberrant chromosome constitution (aneuploidy) of the tumor cells. To facilitate further research on this topic using pattern recognition, we present the first ever publicly available dataset of atypical and normal MFs (AMi-Br). For this, we utilized two of the most popular MF datasets (MIDOG 2021 and TUPAC) and subclassified all MFs using a three expert majority vote. Our final dataset consists of 3,720 MFs, split into 832 AMFs (22.4%) and 2,888 normal MFs (77.6%) across all 223 tumor cases in the combined set. We provide baseline classification experiments to investigate the consistency of the dataset, using a Monte Carlo cross-validation and different strategies to combat class imbalance. We found an averaged balanced accuracy of up to 0.806 when using a patch-level data set split, and up to 0.713 when using a patient-level split.
On the Value of PHH3 for Mitotic Figure Detection on H&E-stained Images
Jun 28, 2024The count of mitotic figures (MFs) observed in hematoxylin and eosin (H&E)-stained slides is an important prognostic marker as it is a measure for tumor cell proliferation. However, the identification of MFs has a known low inter-rater agreement. Deep learning algorithms can standardize this task, but they require large amounts of annotated data for training and validation. Furthermore, label noise introduced during the annotation process may impede the algorithm's performance. Unlike H&E, the mitosis-specific antibody phospho-histone H3 (PHH3) specifically highlights MFs. Counting MFs on slides stained against PHH3 leads to higher agreement among raters and has therefore recently been used as a ground truth for the annotation of MFs in H&E. However, as PHH3 facilitates the recognition of cells indistinguishable from H&E stain alone, the use of this ground truth could potentially introduce noise into the H&E-related dataset, impacting model performance. This study analyzes the impact of PHH3-assisted MF annotation on inter-rater reliability and object level agreement through an extensive multi-rater experiment. We found that the annotators' object-level agreement increased when using PHH3-assisted labeling. Subsequently, MF detectors were evaluated on the resulting datasets to investigate the influence of PHH3-assisted labeling on the models' performance. Additionally, a novel dual-stain MF detector was developed to investigate the interpretation-shift of PHH3-assisted labels used in H&E, which clearly outperformed single-stain detectors. However, the PHH3-assisted labels did not have a positive effect on solely H&E-based models. The high performance of our dual-input detector reveals an information mismatch between the H&E and PHH3-stained images as the cause of this effect.
Automated Volume Corrected Mitotic Index Calculation Through Annotation-Free Deep Learning using Immunohistochemistry as Reference Standard
Nov 15, 2023
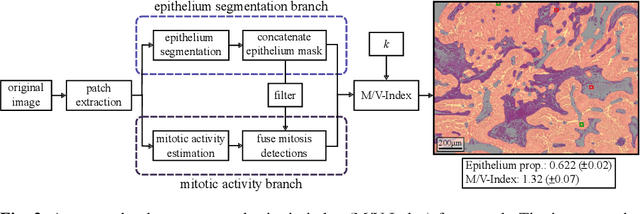
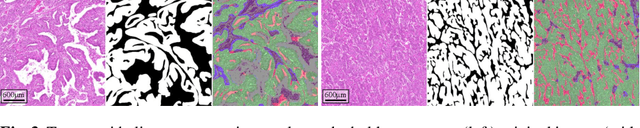
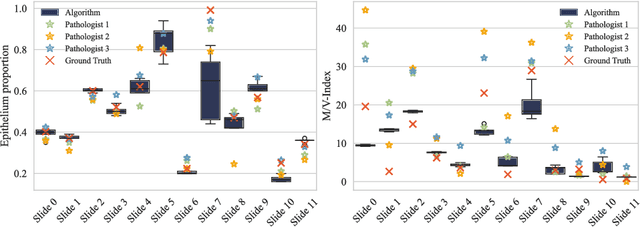
The volume-corrected mitotic index (M/V-Index) was shown to provide prognostic value in invasive breast carcinomas. However, despite its prognostic significance, it is not established as the standard method for assessing aggressive biological behaviour, due to the high additional workload associated with determining the epithelial proportion. In this work, we show that using a deep learning pipeline solely trained with an annotation-free, immunohistochemistry-based approach, provides accurate estimations of epithelial segmentation in canine breast carcinomas. We compare our automatic framework with the manually annotated M/V-Index in a study with three board-certified pathologists. Our results indicate that the deep learning-based pipeline shows expert-level performance, while providing time efficiency and reproducibility.
Domain generalization across tumor types, laboratories, and species -- insights from the 2022 edition of the Mitosis Domain Generalization Challenge
Sep 27, 2023



Recognition of mitotic figures in histologic tumor specimens is highly relevant to patient outcome assessment. This task is challenging for algorithms and human experts alike, with deterioration of algorithmic performance under shifts in image representations. Considerable covariate shifts occur when assessment is performed on different tumor types, images are acquired using different digitization devices, or specimens are produced in different laboratories. This observation motivated the inception of the 2022 challenge on MItosis Domain Generalization (MIDOG 2022). The challenge provided annotated histologic tumor images from six different domains and evaluated the algorithmic approaches for mitotic figure detection provided by nine challenge participants on ten independent domains. Ground truth for mitotic figure detection was established in two ways: a three-expert consensus and an independent, immunohistochemistry-assisted set of labels. This work represents an overview of the challenge tasks, the algorithmic strategies employed by the participants, and potential factors contributing to their success. With an $F_1$ score of 0.764 for the top-performing team, we summarize that domain generalization across various tumor domains is possible with today's deep learning-based recognition pipelines. When assessed against the immunohistochemistry-assisted reference standard, all methods resulted in reduced recall scores, but with only minor changes in the order of participants in the ranking.
Nuclear Morphometry using a Deep Learning-based Algorithm has Prognostic Relevance for Canine Cutaneous Mast Cell Tumors
Sep 26, 2023



Variation in nuclear size and shape is an important criterion of malignancy for many tumor types; however, categorical estimates by pathologists have poor reproducibility. Measurements of nuclear characteristics (morphometry) can improve reproducibility, but manual methods are time consuming. In this study, we evaluated fully automated morphometry using a deep learning-based algorithm in 96 canine cutaneous mast cell tumors with information on patient survival. Algorithmic morphometry was compared with karyomegaly estimates by 11 pathologists, manual nuclear morphometry of 12 cells by 9 pathologists, and the mitotic count as a benchmark. The prognostic value of automated morphometry was high with an area under the ROC curve regarding the tumor-specific survival of 0.943 (95% CI: 0.889 - 0.996) for the standard deviation (SD) of nuclear area, which was higher than manual morphometry of all pathologists combined (0.868, 95% CI: 0.737 - 0.991) and the mitotic count (0.885, 95% CI: 0.765 - 1.00). At the proposed thresholds, the hazard ratio for algorithmic morphometry (SD of nuclear area $\geq 9.0 \mu m^2$) was 18.3 (95% CI: 5.0 - 67.1), for manual morphometry (SD of nuclear area $\geq 10.9 \mu m^2$) 9.0 (95% CI: 6.0 - 13.4), for karyomegaly estimates 7.6 (95% CI: 5.7 - 10.1), and for the mitotic count 30.5 (95% CI: 7.8 - 118.0). Inter-rater reproducibility for karyomegaly estimates was fair ($\kappa$ = 0.226) with highly variable sensitivity/specificity values for the individual pathologists. Reproducibility for manual morphometry (SD of nuclear area) was good (ICC = 0.654). This study supports the use of algorithmic morphometry as a prognostic test to overcome the limitations of estimates and manual measurements.
Deep Learning-Based Automatic Assessment of AgNOR-scores in Histopathology Images
Dec 15, 2022
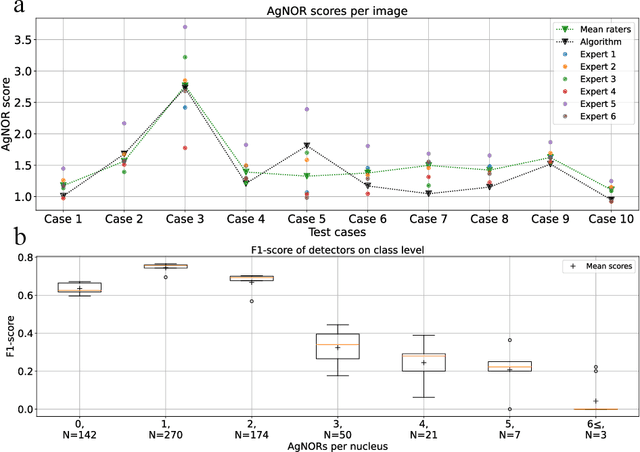
Nucleolar organizer regions (NORs) are parts of the DNA that are involved in RNA transcription. Due to the silver affinity of associated proteins, argyrophilic NORs (AgNORs) can be visualized using silver-based staining. The average number of AgNORs per nucleus has been shown to be a prognostic factor for predicting the outcome of many tumors. Since manual detection of AgNORs is laborious, automation is of high interest. We present a deep learning-based pipeline for automatically determining the AgNOR-score from histopathological sections. An additional annotation experiment was conducted with six pathologists to provide an independent performance evaluation of our approach. Across all raters and images, we found a mean squared error of 0.054 between the AgNOR- scores of the experts and those of the model, indicating that our approach offers performance comparable to humans.
Deep learning-based Subtyping of Atypical and Normal Mitoses using a Hierarchical Anchor-Free Object Detector
Dec 12, 2022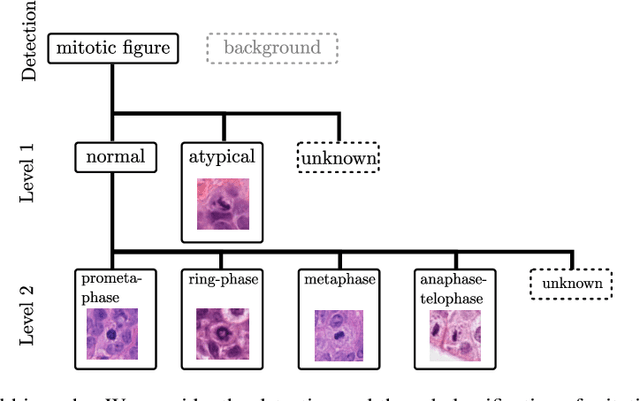
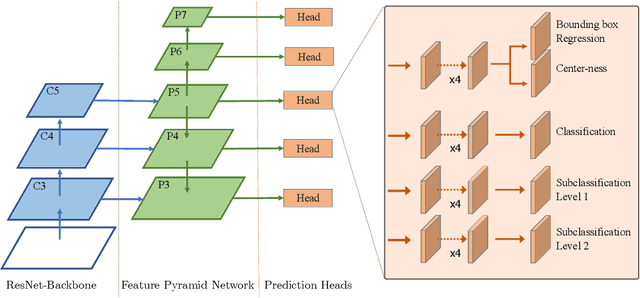
Mitotic activity is key for the assessment of malignancy in many tumors. Moreover, it has been demonstrated that the proportion of abnormal mitosis to normal mitosis is of prognostic significance. Atypical mitotic figures (MF) can be identified morphologically as having segregation abnormalities of the chromatids. In this work, we perform, for the first time, automatic subtyping of mitotic figures into normal and atypical categories according to characteristic morphological appearances of the different phases of mitosis. Using the publicly available MIDOG21 and TUPAC16 breast cancer mitosis datasets, two experts blindly subtyped mitotic figures into five morphological categories. Further, we set up a state-of-the-art object detection pipeline extending the anchor-free FCOS approach with a gated hierarchical subclassification branch. Our labeling experiment indicated that subtyping of mitotic figures is a challenging task and prone to inter-rater disagreement, which we found in 24.89% of MF. Using the more diverse MIDOG21 dataset for training and TUPAC16 for testing, we reached a mean overall average precision score of 0.552, a ROC AUC score of 0.833 for atypical/normal MF and a mean class-averaged ROC-AUC score of 0.977 for discriminating the different phases of cells undergoing mitosis.