 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeArtificial Intelligence-Based Classification of Spitz Tumors
Aug 07, 2025Spitz tumors are diagnostically challenging due to overlap in atypical histological features with conventional melanomas. We investigated to what extent AI models, using histological and/or clinical features, can: (1) distinguish Spitz tumors from conventional melanomas; (2) predict the underlying genetic aberration of Spitz tumors; and (3) predict the diagnostic category of Spitz tumors. The AI models were developed and validated using a dataset of 393 Spitz tumors and 379 conventional melanomas. Predictive performance was measured using the AUROC and the accuracy. The performance of the AI models was compared with that of four experienced pathologists in a reader study. Moreover, a simulation experiment was conducted to investigate the impact of implementing AI-based recommendations for ancillary diagnostic testing on the workflow of the pathology department. The best AI model based on UNI features reached an AUROC of 0.95 and an accuracy of 0.86 in differentiating Spitz tumors from conventional melanomas. The genetic aberration was predicted with an accuracy of 0.55 compared to 0.25 for randomly guessing. The diagnostic category was predicted with an accuracy of 0.51, where random chance-level accuracy equaled 0.33. On all three tasks, the AI models performed better than the four pathologists, although differences were not statistically significant for most individual comparisons. Based on the simulation experiment, implementing AI-based recommendations for ancillary diagnostic testing could reduce material costs, turnaround times, and examinations. In conclusion, the AI models achieved a strong predictive performance in distinguishing between Spitz tumors and conventional melanomas. On the more challenging tasks of predicting the genetic aberration and the diagnostic category of Spitz tumors, the AI models performed better than random chance.
Artificial Intelligence-Based Triaging of Cutaneous Melanocytic Lesions
Oct 14, 2024



Pathologists are facing an increasing workload due to a growing volume of cases and the need for more comprehensive diagnoses. Aiming to facilitate workload reduction and faster turnaround times, we developed an artificial intelligence (AI) model for triaging cutaneous melanocytic lesions based on whole slide images. The AI model was developed and validated using a retrospective cohort from the UMC Utrecht. The dataset consisted of 52,202 whole slide images from 27,167 unique specimens, acquired from 20,707 patients. Specimens with only common nevi were assigned to the low complexity category (86.6%). In contrast, specimens with any other melanocytic lesion subtype, including non-common nevi, melanocytomas, and melanomas, were assigned to the high complexity category (13.4%). The dataset was split on patient level into a development set (80%) and test sets (20%) for independent evaluation. Predictive performance was primarily measured using the area under the receiver operating characteristic curve (AUROC) and the area under the precision-recall curve (AUPRC). A simulation experiment was performed to study the effect of implementing AI-based triaging in the clinic. The AI model reached an AUROC of 0.966 (95% CI, 0.960-0.972) and an AUPRC of 0.857 (95% CI, 0.836-0.877) on the in-distribution test set, and an AUROC of 0.899 (95% CI, 0.860-0.934) and an AUPRC of 0.498 (95% CI, 0.360-0.639) on the out-of-distribution test set. In the simulation experiment, using random case assignment as baseline, AI-based triaging prevented an average of 43.9 (95% CI, 36-55) initial examinations of high complexity cases by general pathologists for every 500 cases. In conclusion, the AI model achieved a strong predictive performance in differentiating between cutaneous melanocytic lesions of high and low complexity. The improvement in workflow efficiency due to AI-based triaging could be substantial.
Domain generalization across tumor types, laboratories, and species -- insights from the 2022 edition of the Mitosis Domain Generalization Challenge
Sep 27, 2023



Recognition of mitotic figures in histologic tumor specimens is highly relevant to patient outcome assessment. This task is challenging for algorithms and human experts alike, with deterioration of algorithmic performance under shifts in image representations. Considerable covariate shifts occur when assessment is performed on different tumor types, images are acquired using different digitization devices, or specimens are produced in different laboratories. This observation motivated the inception of the 2022 challenge on MItosis Domain Generalization (MIDOG 2022). The challenge provided annotated histologic tumor images from six different domains and evaluated the algorithmic approaches for mitotic figure detection provided by nine challenge participants on ten independent domains. Ground truth for mitotic figure detection was established in two ways: a three-expert consensus and an independent, immunohistochemistry-assisted set of labels. This work represents an overview of the challenge tasks, the algorithmic strategies employed by the participants, and potential factors contributing to their success. With an $F_1$ score of 0.764 for the top-performing team, we summarize that domain generalization across various tumor domains is possible with today's deep learning-based recognition pipelines. When assessed against the immunohistochemistry-assisted reference standard, all methods resulted in reduced recall scores, but with only minor changes in the order of participants in the ranking.
Multi-Scanner Canine Cutaneous Squamous Cell Carcinoma Histopathology Dataset
Jan 11, 2023

In histopathology, scanner-induced domain shifts are known to impede the performance of trained neural networks when tested on unseen data. Multi-domain pre-training or dedicated domain-generalization techniques can help to develop domain-agnostic algorithms. For this, multi-scanner datasets with a high variety of slide scanning systems are highly desirable. We present a publicly available multi-scanner dataset of canine cutaneous squamous cell carcinoma histopathology images, composed of 44 samples digitized with five slide scanners. This dataset provides local correspondences between images and thereby isolates the scanner-induced domain shift from other inherent, e.g. morphology-induced domain shifts. To highlight scanner differences, we present a detailed evaluation of color distributions, sharpness, and contrast of the individual scanner subsets. Additionally, to quantify the inherent scanner-induced domain shift, we train a tumor segmentation network on each scanner subset and evaluate the performance both in- and cross-domain. We achieve a class-averaged in-domain intersection over union coefficient of up to 0.86 and observe a cross-domain performance decrease of up to 0.38, which confirms the inherent domain shift of the presented dataset and its negative impact on the performance of deep neural networks.
Mind the Gap: Scanner-induced domain shifts pose challenges for representation learning in histopathology
Nov 29, 2022
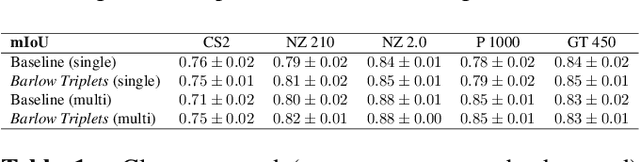
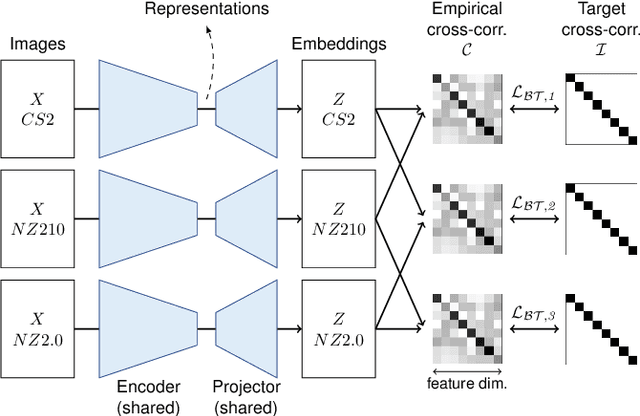
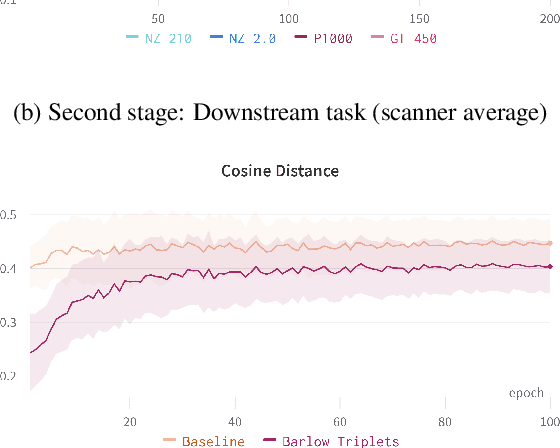
Computer-aided systems in histopathology are often challenged by various sources of domain shift that impact the performance of these algorithms considerably. We investigated the potential of using self-supervised pre-training to overcome scanner-induced domain shifts for the downstream task of tumor segmentation. For this, we present the Barlow Triplets to learn scanner-invariant representations from a multi-scanner dataset with local image correspondences. We show that self-supervised pre-training successfully aligned different scanner representations, which, interestingly only results in a limited benefit for our downstream task. We thereby provide insights into the influence of scanner characteristics for downstream applications and contribute to a better understanding of why established self-supervised methods have not yet shown the same success on histopathology data as they have for natural images.
Mitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022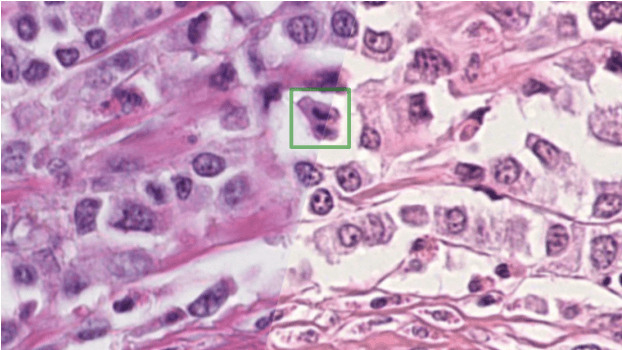
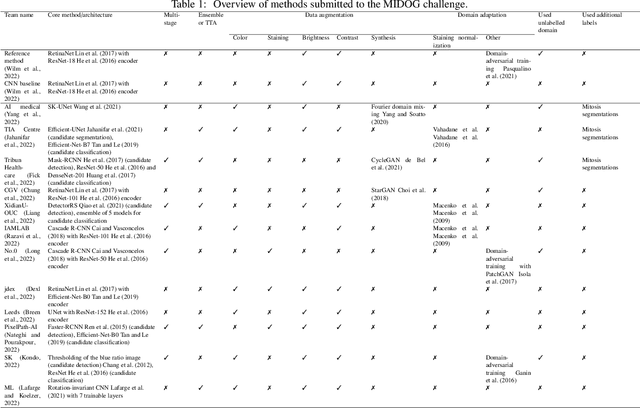
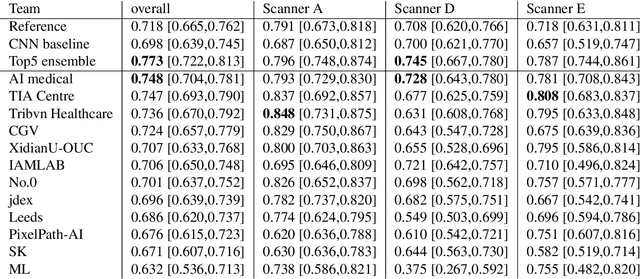
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.
Quantifying the Scanner-Induced Domain Gap in Mitosis Detection
Mar 30, 2021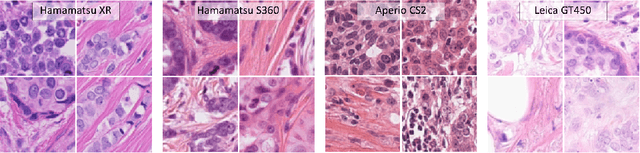

Automated detection of mitotic figures in histopathology images has seen vast improvements, thanks to modern deep learning-based pipelines. Application of these methods, however, is in practice limited by strong variability of images between labs. This results in a domain shift of the images, which causes a performance drop of the models. Hypothesizing that the scanner device plays a decisive role in this effect, we evaluated the susceptibility of a standard mitosis detection approach to the domain shift introduced by using a different whole slide scanner. Our work is based on the MICCAI-MIDOG challenge 2021 data set, which includes 200 tumor cases of human breast cancer and four scanners. Our work indicates that the domain shift induced not by biochemical variability but purely by the choice of acquisition device is underestimated so far. Models trained on images of the same scanner yielded an average F1 score of 0.683, while models trained on a single other scanner only yielded an average F1 score of 0.325. Training on another multi-domain mitosis dataset led to mean F1 scores of 0.52. We found this not to be reflected by domain-shifts measured as proxy A distance-derived metric.
Deep Learning-Based Grading of Ductal Carcinoma In Situ in Breast Histopathology Images
Oct 07, 2020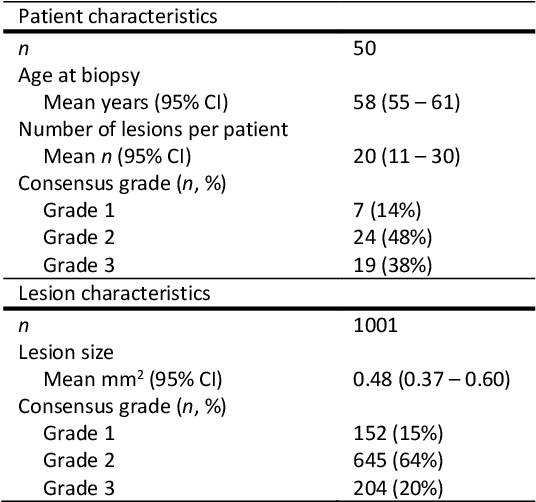
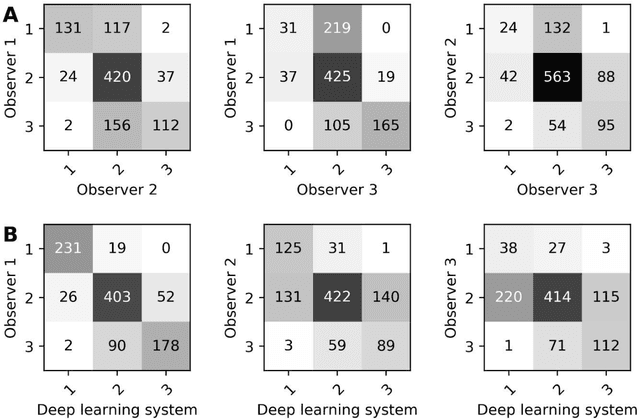
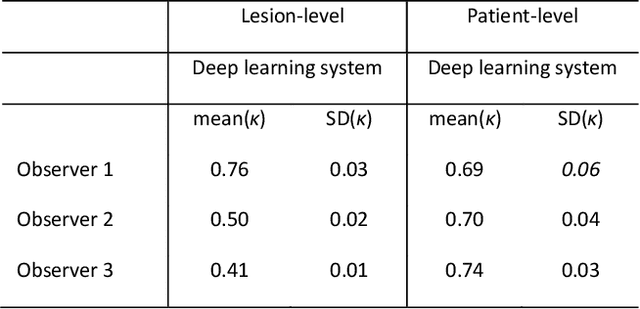
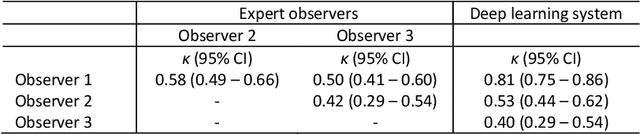
Ductal carcinoma in situ (DCIS) is a non-invasive breast cancer that can progress into invasive ductal carcinoma (IDC). Studies suggest DCIS is often overtreated since a considerable part of DCIS lesions may never progress into IDC. Lower grade lesions have a lower progression speed and risk, possibly allowing treatment de-escalation. However, studies show significant inter-observer variation in DCIS grading. Automated image analysis may provide an objective solution to address high subjectivity of DCIS grading by pathologists. In this study, we developed a deep learning-based DCIS grading system. It was developed using the consensus DCIS grade of three expert observers on a dataset of 1186 DCIS lesions from 59 patients. The inter-observer agreement, measured by quadratic weighted Cohen's kappa, was used to evaluate the system and compare its performance to that of expert observers. We present an analysis of the lesion-level and patient-level inter-observer agreement on an independent test set of 1001 lesions from 50 patients. The deep learning system (dl) achieved on average slightly higher inter-observer agreement to the observers (o1, o2 and o3) ($\kappa_{o1,dl}=0.81, \kappa_{o2,dl}=0.53, \kappa_{o3,dl}=0.40$) than the observers amongst each other ($\kappa_{o1,o2}=0.58, \kappa_{o1,o3}=0.50, \kappa_{o2,o3}=0.42$) at the lesion-level. At the patient-level, the deep learning system achieved similar agreement to the observers ($\kappa_{o1,dl}=0.77, \kappa_{o2,dl}=0.75, \kappa_{o3,dl}=0.70$) as the observers amongst each other ($\kappa_{o1,o2}=0.77, \kappa_{o1,o3}=0.75, \kappa_{o2,o3}=0.72$). In conclusion, we developed a deep learning-based DCIS grading system that achieved a performance similar to expert observers. We believe this is the first automated system that could assist pathologists by providing robust and reproducible second opinions on DCIS grade.
Are pathologist-defined labels reproducible? Comparison of the TUPAC16 mitotic figure dataset with an alternative set of labels
Jul 10, 2020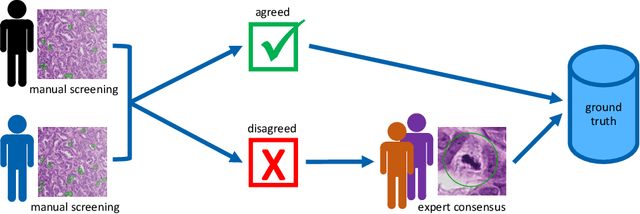
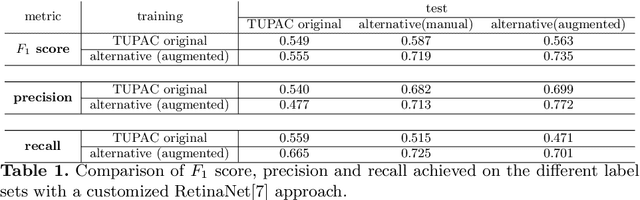
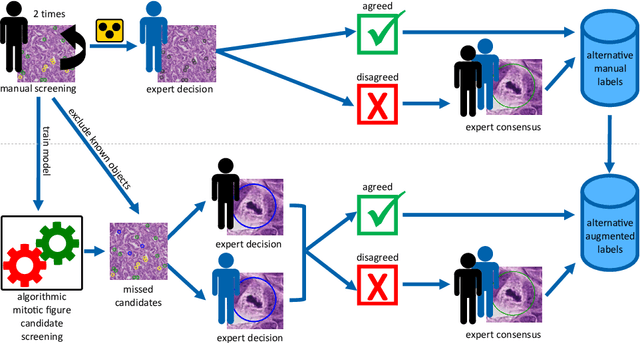
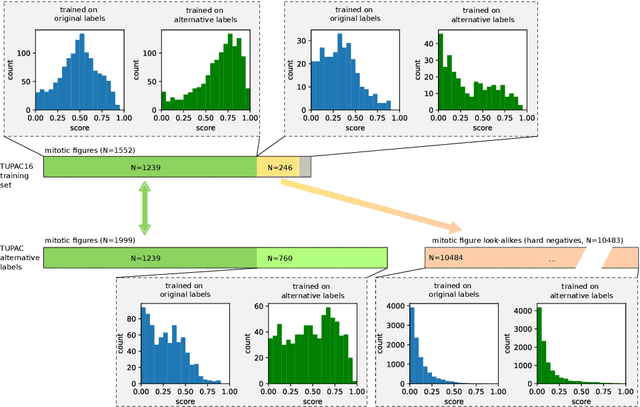
Pathologist-defined labels are the gold standard for histopathological data sets, regardless of well-known limitations in consistency for some tasks. To date, some datasets on mitotic figures are available and were used for development of promising deep learning-based algorithms. In order to assess robustness of those algorithms and reproducibility of their methods it is necessary to test on several independent datasets. The influence of different labeling methods of these available datasets is currently unknown. To tackle this, we present an alternative set of labels for the images of the auxiliary mitosis dataset of the TUPAC16 challenge. Additional to manual mitotic figure screening, we used a novel, algorithm-aided labeling process, that allowed to minimize the risk of missing rare mitotic figures in the images. All potential mitotic figures were independently assessed by two pathologists. The novel, publicly available set of labels contains 1,999 mitotic figures (+28.80%) and additionally includes 10,483 labels of cells with high similarities to mitotic figures (hard examples). We found significant difference comparing F_1 scores between the original label set (0.549) and the new alternative label set (0.735) using a standard deep learning object detection architecture. The models trained on the alternative set showed higher overall confidence values, suggesting a higher overall label consistency. Findings of the present study show that pathologists-defined labels may vary significantly resulting in notable difference in the model performance. Comparison of deep learning-based algorithms between independent datasets with different labeling methods should be done with caution.
Predicting breast tumor proliferation from whole-slide images: the TUPAC16 challenge
Jul 22, 2018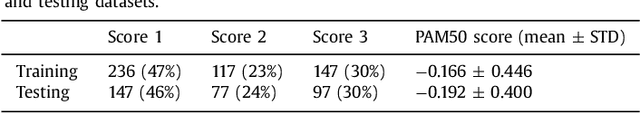
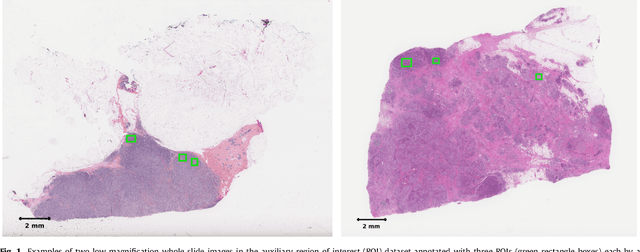
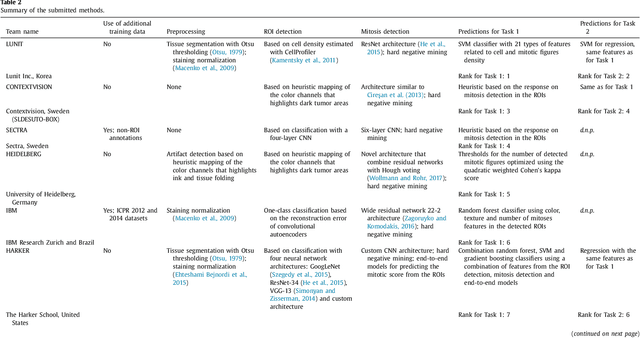
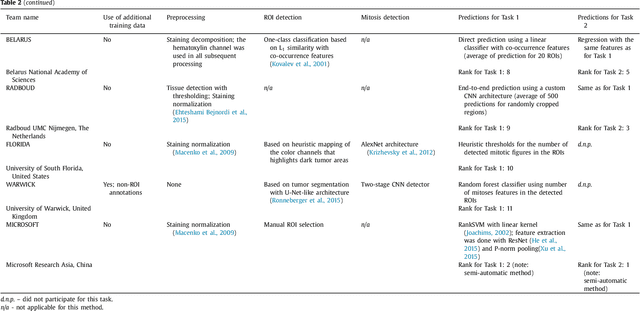
Tumor proliferation is an important biomarker indicative of the prognosis of breast cancer patients. Assessment of tumor proliferation in a clinical setting is highly subjective and labor-intensive task. Previous efforts to automate tumor proliferation assessment by image analysis only focused on mitosis detection in predefined tumor regions. However, in a real-world scenario, automatic mitosis detection should be performed in whole-slide images (WSIs) and an automatic method should be able to produce a tumor proliferation score given a WSI as input. To address this, we organized the TUmor Proliferation Assessment Challenge 2016 (TUPAC16) on prediction of tumor proliferation scores from WSIs. The challenge dataset consisted of 500 training and 321 testing breast cancer histopathology WSIs. In order to ensure fair and independent evaluation, only the ground truth for the training dataset was provided to the challenge participants. The first task of the challenge was to predict mitotic scores, i.e., to reproduce the manual method of assessing tumor proliferation by a pathologist. The second task was to predict the gene expression based PAM50 proliferation scores from the WSI. The best performing automatic method for the first task achieved a quadratic-weighted Cohen's kappa score of $\kappa$ = 0.567, 95% CI [0.464, 0.671] between the predicted scores and the ground truth. For the second task, the predictions of the top method had a Spearman's correlation coefficient of r = 0.617, 95% CI [0.581 0.651] with the ground truth. This was the first study that investigated tumor proliferation assessment from WSIs. The achieved results are promising given the difficulty of the tasks and weakly-labelled nature of the ground truth. However, further research is needed to improve the practical utility of image analysis methods for this task.