 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022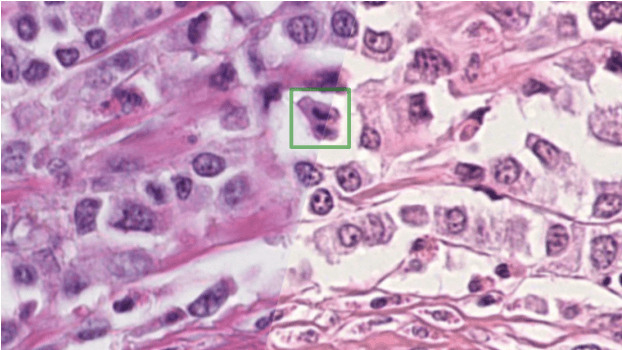
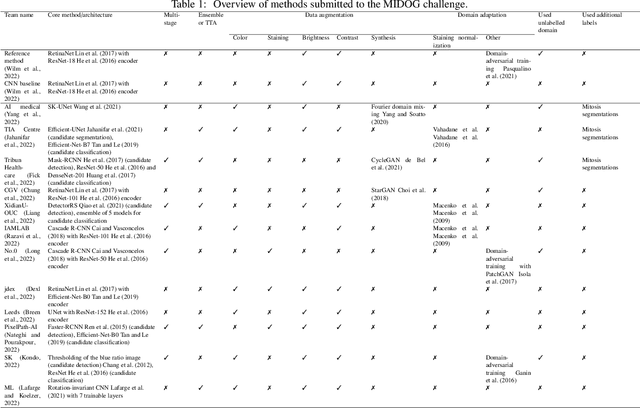
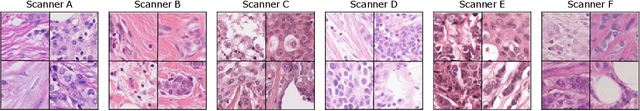
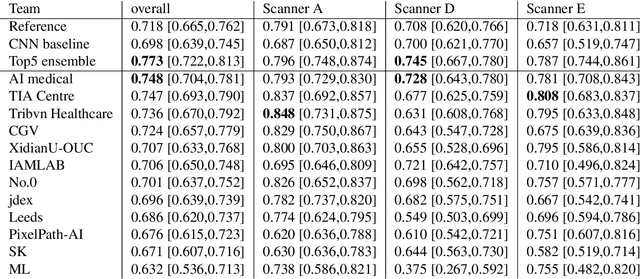
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.
Quantifying the Scanner-Induced Domain Gap in Mitosis Detection
Mar 30, 2021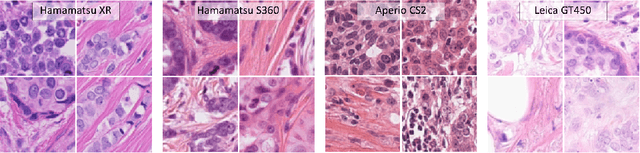

Automated detection of mitotic figures in histopathology images has seen vast improvements, thanks to modern deep learning-based pipelines. Application of these methods, however, is in practice limited by strong variability of images between labs. This results in a domain shift of the images, which causes a performance drop of the models. Hypothesizing that the scanner device plays a decisive role in this effect, we evaluated the susceptibility of a standard mitosis detection approach to the domain shift introduced by using a different whole slide scanner. Our work is based on the MICCAI-MIDOG challenge 2021 data set, which includes 200 tumor cases of human breast cancer and four scanners. Our work indicates that the domain shift induced not by biochemical variability but purely by the choice of acquisition device is underestimated so far. Models trained on images of the same scanner yielded an average F1 score of 0.683, while models trained on a single other scanner only yielded an average F1 score of 0.325. Training on another multi-domain mitosis dataset led to mean F1 scores of 0.52. We found this not to be reflected by domain-shifts measured as proxy A distance-derived metric.