 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-Scanner Canine Cutaneous Squamous Cell Carcinoma Histopathology Dataset
Jan 11, 2023

In histopathology, scanner-induced domain shifts are known to impede the performance of trained neural networks when tested on unseen data. Multi-domain pre-training or dedicated domain-generalization techniques can help to develop domain-agnostic algorithms. For this, multi-scanner datasets with a high variety of slide scanning systems are highly desirable. We present a publicly available multi-scanner dataset of canine cutaneous squamous cell carcinoma histopathology images, composed of 44 samples digitized with five slide scanners. This dataset provides local correspondences between images and thereby isolates the scanner-induced domain shift from other inherent, e.g. morphology-induced domain shifts. To highlight scanner differences, we present a detailed evaluation of color distributions, sharpness, and contrast of the individual scanner subsets. Additionally, to quantify the inherent scanner-induced domain shift, we train a tumor segmentation network on each scanner subset and evaluate the performance both in- and cross-domain. We achieve a class-averaged in-domain intersection over union coefficient of up to 0.86 and observe a cross-domain performance decrease of up to 0.38, which confirms the inherent domain shift of the presented dataset and its negative impact on the performance of deep neural networks.
Mind the Gap: Scanner-induced domain shifts pose challenges for representation learning in histopathology
Nov 29, 2022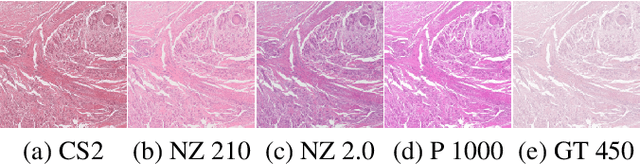
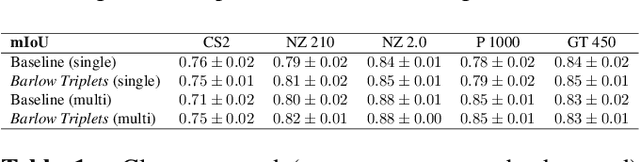
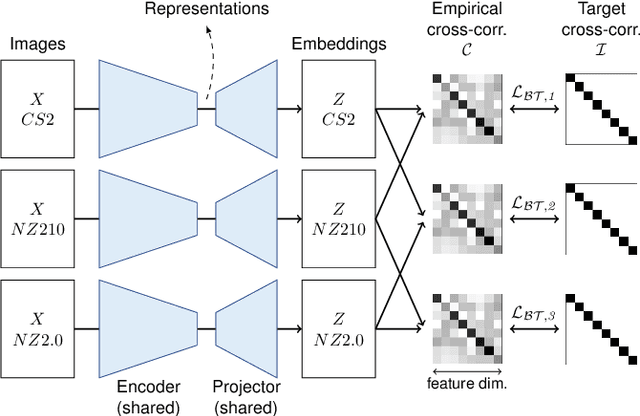
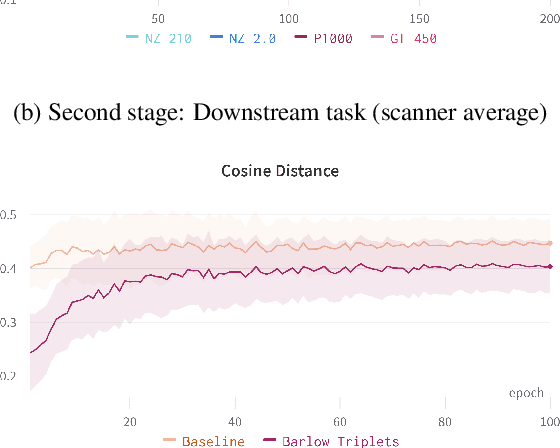
Computer-aided systems in histopathology are often challenged by various sources of domain shift that impact the performance of these algorithms considerably. We investigated the potential of using self-supervised pre-training to overcome scanner-induced domain shifts for the downstream task of tumor segmentation. For this, we present the Barlow Triplets to learn scanner-invariant representations from a multi-scanner dataset with local image correspondences. We show that self-supervised pre-training successfully aligned different scanner representations, which, interestingly only results in a limited benefit for our downstream task. We thereby provide insights into the influence of scanner characteristics for downstream applications and contribute to a better understanding of why established self-supervised methods have not yet shown the same success on histopathology data as they have for natural images.
Pan-Tumor CAnine cuTaneous Cancer Histology (CATCH) Dataset
Jan 27, 2022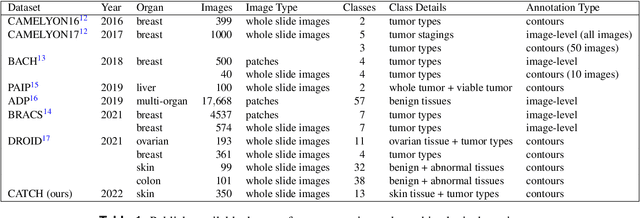
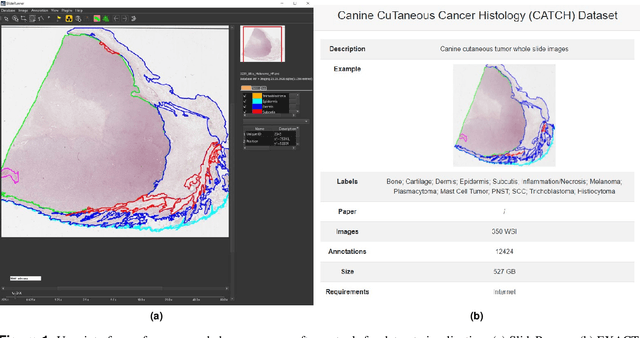
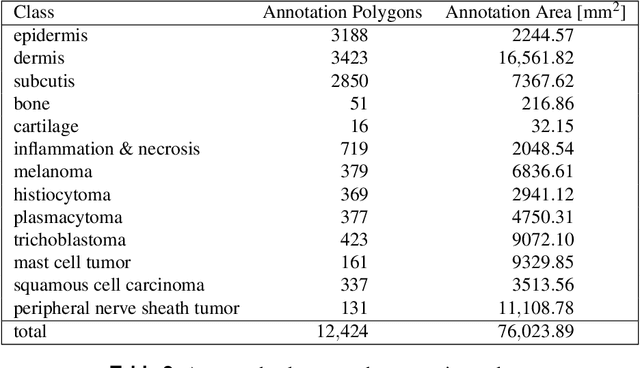

Due to morphological similarities, the differentiation of histologic sections of cutaneous tumors into individual subtypes can be challenging. Recently, deep learning-based approaches have proven their potential for supporting pathologists in this regard. However, many of these supervised algorithms require a large amount of annotated data for robust development. We present a publicly available dataset consisting of 350 whole slide images of seven different canine cutaneous tumors complemented by 12,424 polygon annotations for 13 histologic classes including seven cutaneous tumor subtypes. Regarding sample size and annotation extent, this exceeds most publicly available datasets which are oftentimes limited to the tumor area or merely provide patch-level annotations. We validated our model for tissue segmentation, achieving a class-averaged Jaccard coefficient of 0.7047, and 0.9044 for tumor in particular. For tumor subtype classification, we achieve a slide-level accuracy of 0.9857. Since canine cutaneous tumors possess various histologic homologies to human tumors, we believe that the added value of this dataset is not limited to veterinary pathology but extends to more general fields of application.
Dataset on Bi- and Multi-Nucleated Tumor Cells in Canine Cutaneous Mast Cell Tumors
Jan 05, 2021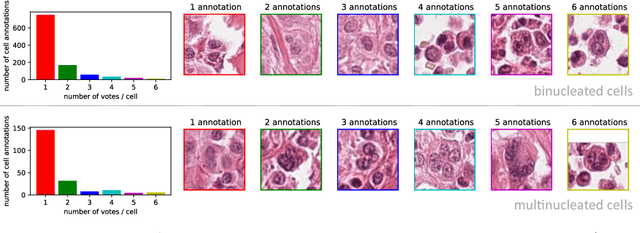
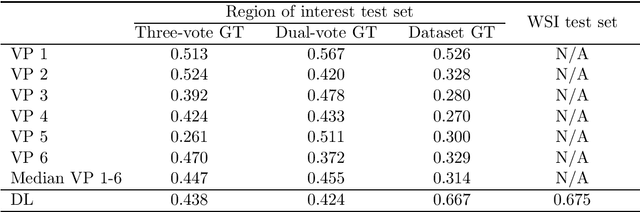
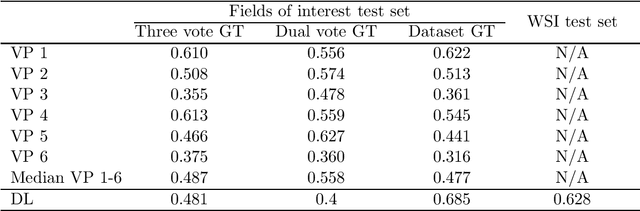
Tumor cells with two nuclei (binucleated cells, BiNC) or more nuclei (multinucleated cells, MuNC) indicate an increased amount of cellular genetic material which is thought to facilitate oncogenesis, tumor progression and treatment resistance. In canine cutaneous mast cell tumors (ccMCT), binucleation and multinucleation are parameters used in cytologic and histologic grading schemes (respectively) which correlate with poor patient outcome. For this study, we created the first open source data-set with 19,983 annotations of BiNC and 1,416 annotations of MuNC in 32 histological whole slide images of ccMCT. Labels were created by a pathologist and an algorithmic-aided labeling approach with expert review of each generated candidate. A state-of-the-art deep learning-based model yielded an $F_1$ score of 0.675 for BiNC and 0.623 for MuNC on 11 test whole slide images. In regions of interest ($2.37 mm^2$) extracted from these test images, 6 pathologists had an object detection performance between 0.270 - 0.526 for BiNC and 0.316 - 0.622 for MuNC, while our model archived an $F_1$ score of 0.667 for BiNC and 0.685 for MuNC. This open dataset can facilitate development of automated image analysis for this task and may thereby help to promote standardization of this facet of histologic tumor prognostication.
Field of Interest Prediction for Computer-Aided Mitotic Count
Feb 12, 2019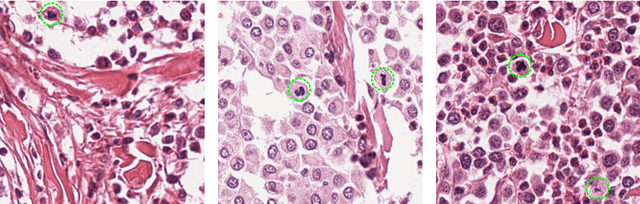
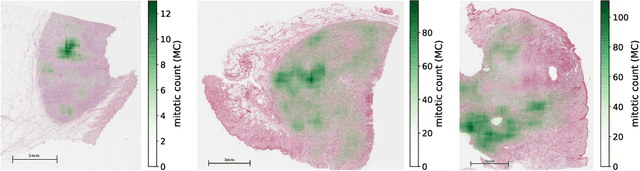
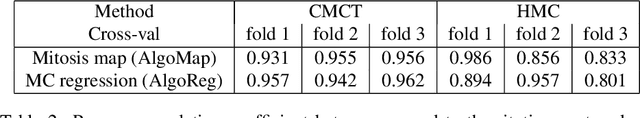
Manual counts of mitotic figures, which are determined in the tumor region with the highest mitotic activity, are a key parameter of most tumor grading schemes. It is however strongly dependent on the area selection. To reduce potential variability of prognosis due to this, we propose to use an algorithmic field of interest prediction to assess the area of highest mitotic activity in a whole-slide image. Methods: We evaluated two state-of-the-art methods, all based on the use of deep convolutional neural networks on their ability to predict the mitotic count in digital histopathology slides. We evaluated them on a novel dataset of 32 completely annotated whole slide images from canine cutaneous mast cell tumors (CMCT) and one publicly available human mamma carcinoma (HMC) dataset. We first compared the mitotic counts (MC) predicted by the two models with the ground truth MC on both data sets. Second, for the CMCT data set, we compared the computationally predicted position and MC of the area of highest mitotic activity with size-equivalent areas selected by eight veterinary pathologists. Results: We found a high correlation between the mitotic count as predicted by the models (Pearson's correlation coefficient between 0.931 and 0.962 for the CMCT data set and between 0.801 and 0.986 for the HMC data set) on the slides. For the CMCT data set, this is also reflected in the predicted position representing mitotic counts in mostly the upper quartile of the slide's ground truth MC distribution. Further, we found strong differences between experts in position selection. Conclusion: While the mitotic counts in areas selected by the experts substantially varied, both algorithmic approaches were consistently able to generate a good estimate of the area of highest mitotic count. To achieve better inter-rater agreement, we propose to use computer-based area selection for manual mitotic count.