 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIs Self-Supervision Enough? Benchmarking Foundation Models Against End-to-End Training for Mitotic Figure Classification
Dec 09, 2024


Foundation models (FMs), i.e., models trained on a vast amount of typically unlabeled data, have become popular and available recently for the domain of histopathology. The key idea is to extract semantically rich vectors from any input patch, allowing for the use of simple subsequent classification networks potentially reducing the required amounts of labeled data, and increasing domain robustness. In this work, we investigate to which degree this also holds for mitotic figure classification. Utilizing two popular public mitotic figure datasets, we compared linear probing of five publicly available FMs against models trained on ImageNet and a simple ResNet50 end-to-end-trained baseline. We found that the end-to-end-trained baseline outperformed all FM-based classifiers, regardless of the amount of data provided. Additionally, we did not observe the FM-based classifiers to be more robust against domain shifts, rendering both of the above assumptions incorrect.
On the Value of PHH3 for Mitotic Figure Detection on H&E-stained Images
Jun 28, 2024The count of mitotic figures (MFs) observed in hematoxylin and eosin (H&E)-stained slides is an important prognostic marker as it is a measure for tumor cell proliferation. However, the identification of MFs has a known low inter-rater agreement. Deep learning algorithms can standardize this task, but they require large amounts of annotated data for training and validation. Furthermore, label noise introduced during the annotation process may impede the algorithm's performance. Unlike H&E, the mitosis-specific antibody phospho-histone H3 (PHH3) specifically highlights MFs. Counting MFs on slides stained against PHH3 leads to higher agreement among raters and has therefore recently been used as a ground truth for the annotation of MFs in H&E. However, as PHH3 facilitates the recognition of cells indistinguishable from H&E stain alone, the use of this ground truth could potentially introduce noise into the H&E-related dataset, impacting model performance. This study analyzes the impact of PHH3-assisted MF annotation on inter-rater reliability and object level agreement through an extensive multi-rater experiment. We found that the annotators' object-level agreement increased when using PHH3-assisted labeling. Subsequently, MF detectors were evaluated on the resulting datasets to investigate the influence of PHH3-assisted labeling on the models' performance. Additionally, a novel dual-stain MF detector was developed to investigate the interpretation-shift of PHH3-assisted labels used in H&E, which clearly outperformed single-stain detectors. However, the PHH3-assisted labels did not have a positive effect on solely H&E-based models. The high performance of our dual-input detector reveals an information mismatch between the H&E and PHH3-stained images as the cause of this effect.
Model-based Cleaning of the QUILT-1M Pathology Dataset for Text-Conditional Image Synthesis
Apr 11, 2024The QUILT-1M dataset is the first openly available dataset containing images harvested from various online sources. While it provides a huge data variety, the image quality and composition is highly heterogeneous, impacting its utility for text-conditional image synthesis. We propose an automatic pipeline that provides predictions of the most common impurities within the images, e.g., visibility of narrators, desktop environment and pathology software, or text within the image. Additionally, we propose to use semantic alignment filtering of the image-text pairs. Our findings demonstrate that by rigorously filtering the dataset, there is a substantial enhancement of image fidelity in text-to-image tasks.
Re-identification from histopathology images
Mar 19, 2024



In numerous studies, deep learning algorithms have proven their potential for the analysis of histopathology images, for example, for revealing the subtypes of tumors or the primary origin of metastases. These models require large datasets for training, which must be anonymized to prevent possible patient identity leaks. This study demonstrates that even relatively simple deep learning algorithms can re-identify patients in large histopathology datasets with substantial accuracy. We evaluated our algorithms on two TCIA datasets including lung squamous cell carcinoma (LSCC) and lung adenocarcinoma (LUAD). We also demonstrate the algorithm's performance on an in-house dataset of meningioma tissue. We predicted the source patient of a slide with F1 scores of 50.16 % and 52.30 % on the LSCC and LUAD datasets, respectively, and with 62.31 % on our meningioma dataset. Based on our findings, we formulated a risk assessment scheme to estimate the risk to the patient's privacy prior to publication.
Deep Learning model predicts the c-Kit-11 mutational status of canine cutaneous mast cell tumors by HE stained histological slides
Jan 02, 2024
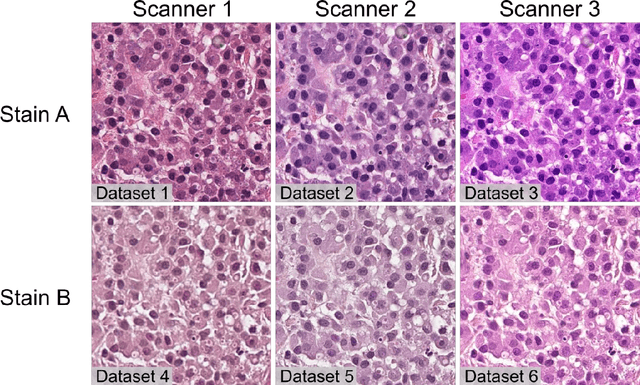
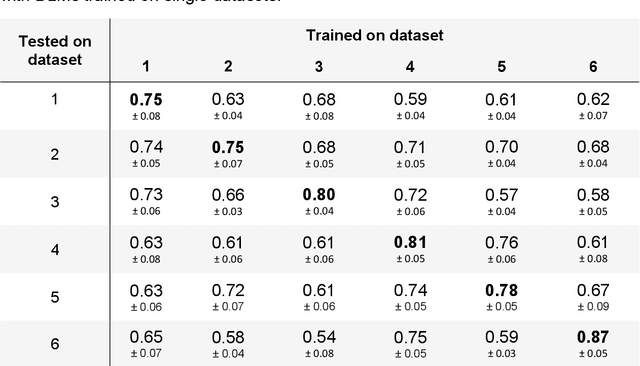
Numerous prognostic factors are currently assessed histopathologically in biopsies of canine mast cell tumors to evaluate clinical behavior. In addition, PCR analysis of the c-Kit exon 11 mutational status is often performed to evaluate the potential success of a tyrosine kinase inhibitor therapy. This project aimed at training deep learning models (DLMs) to identify the c-Kit-11 mutational status of MCTs solely based on morphology without additional molecular analysis. HE slides of 195 mutated and 173 non-mutated tumors were stained consecutively in two different laboratories and scanned with three different slide scanners. This resulted in six different datasets (stain-scanner variations) of whole slide images. DLMs were trained with single and mixed datasets and their performances was assessed under scanner and staining domain shifts. The DLMs correctly classified HE slides according to their c-Kit 11 mutation status in, on average, 87% of cases for the best-suited stain-scanner variant. A relevant performance drop could be observed when the stain-scanner combination of the training and test dataset differed. Multi-variant datasets improved the average accuracy but did not reach the maximum accuracy of algorithms trained and tested on the same stain-scanner variant. In summary, DLM-assisted morphological examination of MCTs can predict c-Kit-exon 11 mutational status of MCTs with high accuracy. However, the recognition performance is impeded by a change of scanner or staining protocol. Larger data sets with higher numbers of scans originating from different laboratories and scanners may lead to more robust DLMs to identify c-Kit mutations in HE slides.
Automated Volume Corrected Mitotic Index Calculation Through Annotation-Free Deep Learning using Immunohistochemistry as Reference Standard
Nov 15, 2023
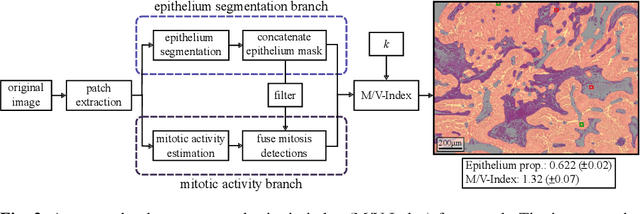
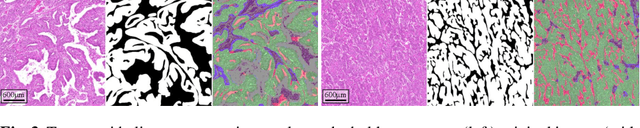
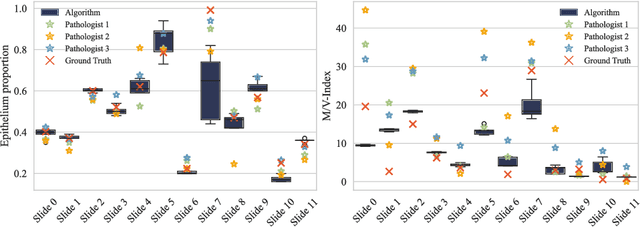
The volume-corrected mitotic index (M/V-Index) was shown to provide prognostic value in invasive breast carcinomas. However, despite its prognostic significance, it is not established as the standard method for assessing aggressive biological behaviour, due to the high additional workload associated with determining the epithelial proportion. In this work, we show that using a deep learning pipeline solely trained with an annotation-free, immunohistochemistry-based approach, provides accurate estimations of epithelial segmentation in canine breast carcinomas. We compare our automatic framework with the manually annotated M/V-Index in a study with three board-certified pathologists. Our results indicate that the deep learning-based pipeline shows expert-level performance, while providing time efficiency and reproducibility.
Few Shot Learning for the Classification of Confocal Laser Endomicroscopy Images of Head and Neck Tumors
Nov 13, 2023


The surgical removal of head and neck tumors requires safe margins, which are usually confirmed intraoperatively by means of frozen sections. This method is, in itself, an oversampling procedure, which has a relatively low sensitivity compared to the definitive tissue analysis on paraffin-embedded sections. Confocal laser endomicroscopy (CLE) is an in-vivo imaging technique that has shown its potential in the live optical biopsy of tissue. An automated analysis of this notoriously difficult to interpret modality would help surgeons. However, the images of CLE show a wide variability of patterns, caused both by individual factors but also, and most strongly, by the anatomical structures of the imaged tissue, making it a challenging pattern recognition task. In this work, we evaluate four popular few shot learning (FSL) methods towards their capability of generalizing to unseen anatomical domains in CLE images. We evaluate this on images of sinunasal tumors (SNT) from five patients and on images of the vocal folds (VF) from 11 patients using a cross-validation scheme. The best respective approach reached a median accuracy of 79.6% on the rather homogeneous VF dataset, but only of 61.6% for the highly diverse SNT dataset. Our results indicate that FSL on CLE images is viable, but strongly affected by the number of patients, as well as the diversity of anatomical patterns.
Domain generalization across tumor types, laboratories, and species -- insights from the 2022 edition of the Mitosis Domain Generalization Challenge
Sep 27, 2023



Recognition of mitotic figures in histologic tumor specimens is highly relevant to patient outcome assessment. This task is challenging for algorithms and human experts alike, with deterioration of algorithmic performance under shifts in image representations. Considerable covariate shifts occur when assessment is performed on different tumor types, images are acquired using different digitization devices, or specimens are produced in different laboratories. This observation motivated the inception of the 2022 challenge on MItosis Domain Generalization (MIDOG 2022). The challenge provided annotated histologic tumor images from six different domains and evaluated the algorithmic approaches for mitotic figure detection provided by nine challenge participants on ten independent domains. Ground truth for mitotic figure detection was established in two ways: a three-expert consensus and an independent, immunohistochemistry-assisted set of labels. This work represents an overview of the challenge tasks, the algorithmic strategies employed by the participants, and potential factors contributing to their success. With an $F_1$ score of 0.764 for the top-performing team, we summarize that domain generalization across various tumor domains is possible with today's deep learning-based recognition pipelines. When assessed against the immunohistochemistry-assisted reference standard, all methods resulted in reduced recall scores, but with only minor changes in the order of participants in the ranking.
Nuclear Morphometry using a Deep Learning-based Algorithm has Prognostic Relevance for Canine Cutaneous Mast Cell Tumors
Sep 26, 2023Variation in nuclear size and shape is an important criterion of malignancy for many tumor types; however, categorical estimates by pathologists have poor reproducibility. Measurements of nuclear characteristics (morphometry) can improve reproducibility, but manual methods are time consuming. In this study, we evaluated fully automated morphometry using a deep learning-based algorithm in 96 canine cutaneous mast cell tumors with information on patient survival. Algorithmic morphometry was compared with karyomegaly estimates by 11 pathologists, manual nuclear morphometry of 12 cells by 9 pathologists, and the mitotic count as a benchmark. The prognostic value of automated morphometry was high with an area under the ROC curve regarding the tumor-specific survival of 0.943 (95% CI: 0.889 - 0.996) for the standard deviation (SD) of nuclear area, which was higher than manual morphometry of all pathologists combined (0.868, 95% CI: 0.737 - 0.991) and the mitotic count (0.885, 95% CI: 0.765 - 1.00). At the proposed thresholds, the hazard ratio for algorithmic morphometry (SD of nuclear area $\geq 9.0 \mu m^2$) was 18.3 (95% CI: 5.0 - 67.1), for manual morphometry (SD of nuclear area $\geq 10.9 \mu m^2$) 9.0 (95% CI: 6.0 - 13.4), for karyomegaly estimates 7.6 (95% CI: 5.7 - 10.1), and for the mitotic count 30.5 (95% CI: 7.8 - 118.0). Inter-rater reproducibility for karyomegaly estimates was fair ($\kappa$ = 0.226) with highly variable sensitivity/specificity values for the individual pathologists. Reproducibility for manual morphometry (SD of nuclear area) was good (ICC = 0.654). This study supports the use of algorithmic morphometry as a prognostic test to overcome the limitations of estimates and manual measurements.
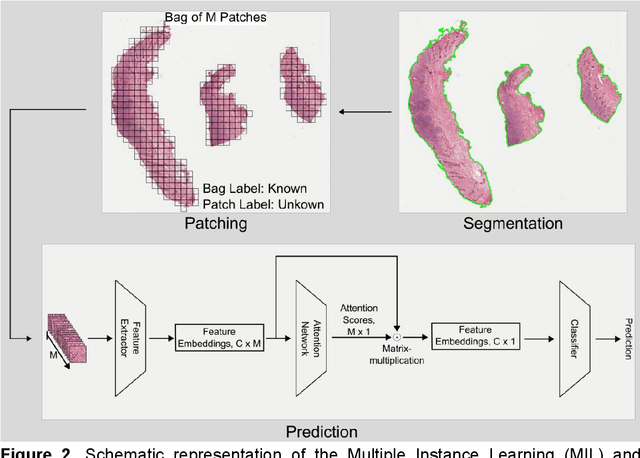
Attention-based Multiple Instance Learning for Survival Prediction on Lung Cancer Tissue Microarrays
Dec 15, 2022

Attention-based multiple instance learning (AMIL) algorithms have proven to be successful in utilizing gigapixel whole-slide images (WSIs) for a variety of different computational pathology tasks such as outcome prediction and cancer subtyping problems. We extended an AMIL approach to the task of survival prediction by utilizing the classical Cox partial likelihood as a loss function, converting the AMIL model into a nonlinear proportional hazards model. We applied the model to tissue microarray (TMA) slides of 330 lung cancer patients. The results show that AMIL approaches can handle very small amounts of tissue from a TMA and reach similar C-index performance compared to established survival prediction methods trained with highly discriminative clinical factors such as age, cancer grade, and cancer stage