 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRethinking U-net Skip Connections for Biomedical Image Segmentation
Feb 13, 2024The U-net architecture has significantly impacted deep learning-based segmentation of medical images. Through the integration of long-range skip connections, it facilitated the preservation of high-resolution features. Out-of-distribution data can, however, substantially impede the performance of neural networks. Previous works showed that the trained network layers differ in their susceptibility to this domain shift, e.g., shallow layers are more affected than deeper layers. In this work, we investigate the implications of this observation of layer sensitivity to domain shifts of U-net-style segmentation networks. By copying features of shallow layers to corresponding decoder blocks, these bear the risk of re-introducing domain-specific information. We used a synthetic dataset to model different levels of data distribution shifts and evaluated the impact on downstream segmentation performance. We quantified the inherent domain susceptibility of each network layer, using the Hellinger distance. These experiments confirmed the higher domain susceptibility of earlier network layers. When gradually removing skip connections, a decrease in domain susceptibility of deeper layers could be observed. For downstream segmentation performance, the original U-net outperformed the variant without any skip connections. The best performance, however, was achieved when removing the uppermost skip connection - not only in the presence of domain shifts but also for in-domain test data. We validated our results on three clinical datasets - two histopathology datasets and one magnetic resonance dataset - with performance increases of up to 10% in-domain and 13% cross-domain when removing the uppermost skip connection.
Deep learning-based Subtyping of Atypical and Normal Mitoses using a Hierarchical Anchor-Free Object Detector
Dec 12, 2022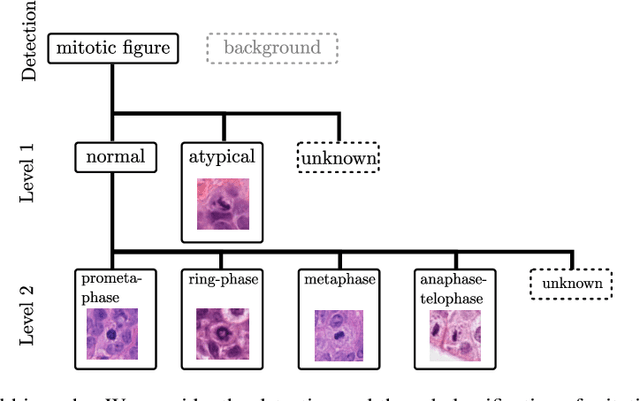
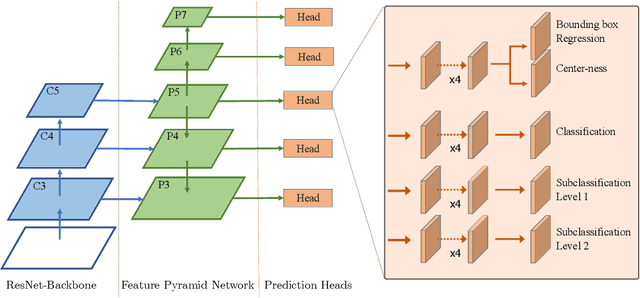
Mitotic activity is key for the assessment of malignancy in many tumors. Moreover, it has been demonstrated that the proportion of abnormal mitosis to normal mitosis is of prognostic significance. Atypical mitotic figures (MF) can be identified morphologically as having segregation abnormalities of the chromatids. In this work, we perform, for the first time, automatic subtyping of mitotic figures into normal and atypical categories according to characteristic morphological appearances of the different phases of mitosis. Using the publicly available MIDOG21 and TUPAC16 breast cancer mitosis datasets, two experts blindly subtyped mitotic figures into five morphological categories. Further, we set up a state-of-the-art object detection pipeline extending the anchor-free FCOS approach with a gated hierarchical subclassification branch. Our labeling experiment indicated that subtyping of mitotic figures is a challenging task and prone to inter-rater disagreement, which we found in 24.89% of MF. Using the more diverse MIDOG21 dataset for training and TUPAC16 for testing, we reached a mean overall average precision score of 0.552, a ROC AUC score of 0.833 for atypical/normal MF and a mean class-averaged ROC-AUC score of 0.977 for discriminating the different phases of cells undergoing mitosis.
Enabling Collagen Quantification on HE-stained Slides Through Stain Deconvolution and Restained HE-HES
Nov 17, 2022



In histology, the presence of collagen in the extra-cellular matrix has both diagnostic and prognostic value for cancer malignancy, and can be highlighted by adding Saffron (S) to a routine Hematoxylin and Eosin (HE) staining. However, Saffron is not usually added because of the additional cost and because pathologists are accustomed to HE, with the exception of France-based laboratories. In this paper, we show that it is possible to quantify the collagen content from the HE image alone and to digitally create an HES image. To do so, we trained a UNet to predict the Saffron densities from HE images. We created a dataset of registered, restained HE-HES slides and we extracted the Saffron concentrations as ground truth using stain deconvolution on the HES images. Our model reached a Mean Absolute Error of 0.0668 $\pm$ 0.0002 (Saffron values between 0 and 1) on a 3-fold testing set. We hope our approach can aid in improving the clinical workflow while reducing reagent costs for laboratories.
Interpretable HER2 scoring by evaluating clinical Guidelines through a weakly supervised, constrained Deep Learning Approach
Nov 17, 2022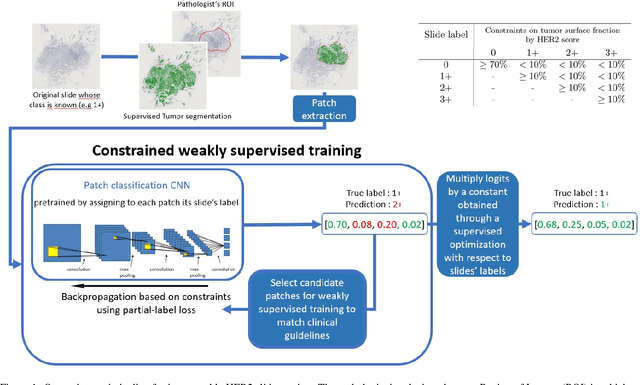

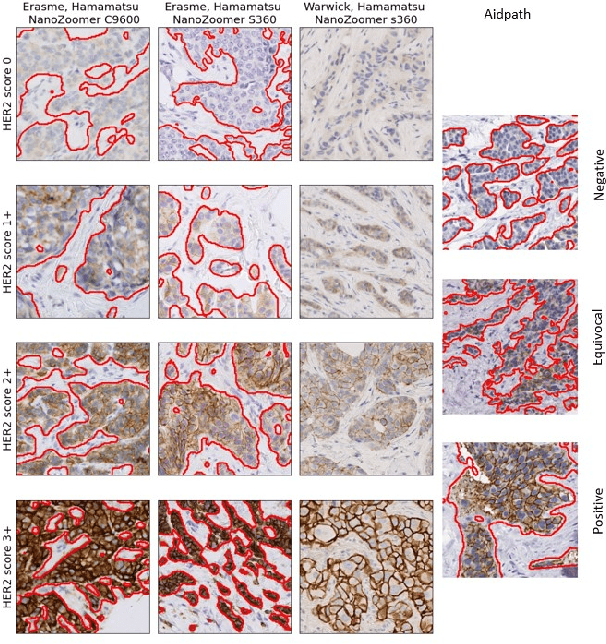
The evaluation of the Human Epidermal growth factor Receptor-2 (HER2) expression is an important prognostic biomarker for breast cancer treatment selection. However, HER2 scoring has notoriously high interobserver variability due to stain variations between centers and the need to estimate visually the staining intensity in specific percentages of tumor area. In this paper, focusing on the interpretability of HER2 scoring by a pathologist, we propose a semi-automatic, two-stage deep learning approach that directly evaluates the clinical HER2 guidelines defined by the American Society of Clinical Oncology/ College of American Pathologists (ASCO/CAP). In the first stage, we segment the invasive tumor over the user-indicated Region of Interest (ROI). Then, in the second stage, we classify the tumor tissue into four HER2 classes. For the classification stage, we use weakly supervised, constrained optimization to find a model that classifies cancerous patches such that the tumor surface percentage meets the guidelines specification of each HER2 class. We end the second stage by freezing the model and refining its output logits in a supervised way to all slide labels in the training set. To ensure the quality of our dataset's labels, we conducted a multi-pathologist HER2 scoring consensus. For the assessment of doubtful cases where no consensus was found, our model can help by interpreting its HER2 class percentages output. We achieve a performance of 0.78 in F1-score on the test set while keeping our model interpretable for the pathologist, hopefully contributing to interpretable AI models in digital pathology.
Mitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022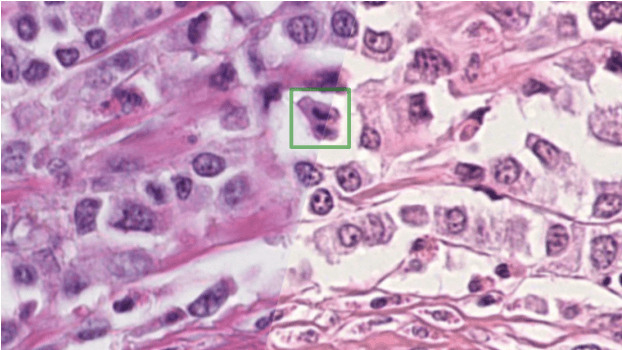
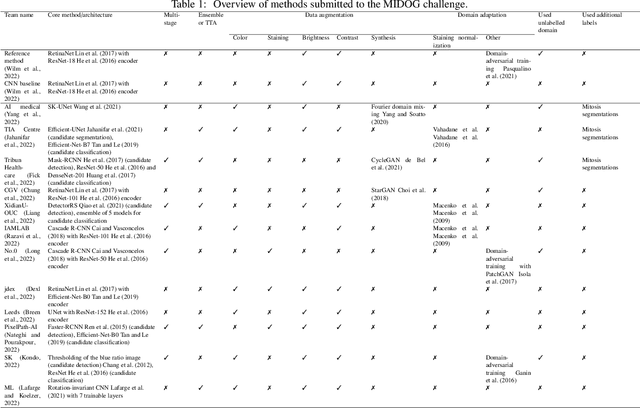
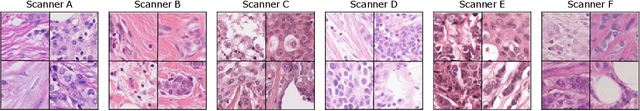
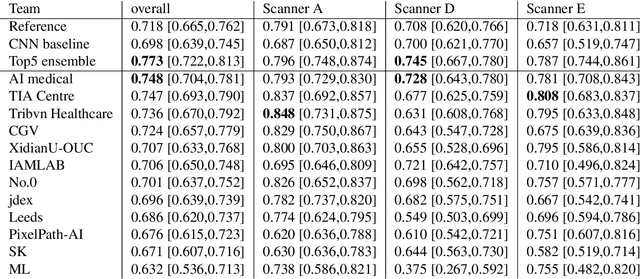
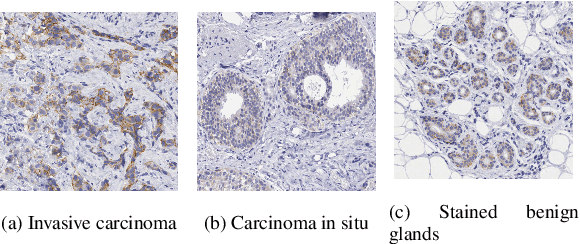
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.
Robust Mitosis Detection Using a Cascade Mask-RCNN Approach With Domain-Specific Residual Cycle-GAN Data Augmentation
Sep 28, 2021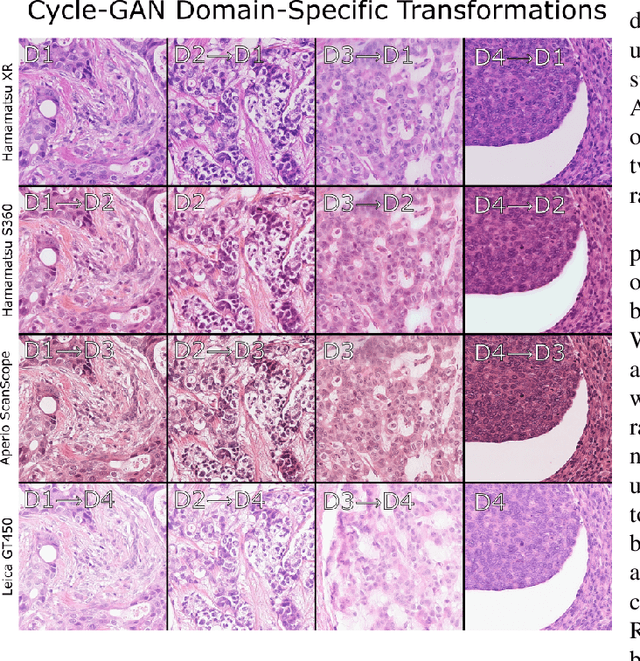
For the MIDOG mitosis detection challenge, we created a cascade algorithm consisting of a Mask-RCNN detector, followed by a classification ensemble consisting of ResNet50 and DenseNet201 to refine detected mitotic candidates. The MIDOG training data consists of 200 frames originating from four scanners, three of which are annotated for mitotic instances with centroid annotations. Our main algorithmic choices are as follows: first, to enhance the generalizability of our detector and classification networks, we use a state-of-the-art residual Cycle-GAN to transform each scanner domain to every other scanner domain. During training, we then randomly load, for each image, one of the four domains. In this way, our networks can learn from the fourth non-annotated scanner domain even if we don't have annotations for it. Second, for training the detector network, rather than using centroid-based fixed-size bounding boxes, we create mitosis-specific bounding boxes. We do this by manually annotating a small selection of mitoses, training a Mask-RCNN on this small dataset, and applying it to the rest of the data to obtain full annotations. We trained the follow-up classification ensemble using only the challenge-provided positive and hard-negative examples. On the preliminary test set, the algorithm scores an F1 score of 0.7578, putting us as the second-place team on the leaderboard.