 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultiST: A Cross-Attention-Based Multimodal Model for Spatial Transcriptomic
Jan 19, 2026Spatial transcriptomics (ST) enables transcriptome-wide profiling while preserving the spatial context of tissues, offering unprecedented opportunities to study tissue organization and cell-cell interactions in situ. Despite recent advances, existing methods often lack effective integration of histological morphology with molecular profiles, relying on shallow fusion strategies or omitting tissue images altogether, which limits their ability to resolve ambiguous spatial domain boundaries. To address this challenge, we propose MultiST, a unified multimodal framework that jointly models spatial topology, gene expression, and tissue morphology through cross-attention-based fusion. MultiST employs graph-based gene encoders with adversarial alignment to learn robust spatial representations, while integrating color-normalized histological features to capture molecular-morphological dependencies and refine domain boundaries. We evaluated the proposed method on 13 diverse ST datasets spanning two organs, including human brain cortex and breast cancer tissue. MultiST yields spatial domains with clearer and more coherent boundaries than existing methods, leading to more stable pseudotime trajectories and more biologically interpretable cell-cell interaction patterns. The MultiST framework and source code are available at https://github.com/LabJunBMI/MultiST.git.
PepEDiff: Zero-Shot Peptide Binder Design via Protein Embedding Diffusion
Jan 19, 2026We present PepEDiff, a novel peptide binder generator that designs binding sequences given a target receptor protein sequence and its pocket residues. Peptide binder generation is critical in therapeutic and biochemical applications, yet many existing methods rely heavily on intermediate structure prediction, adding complexity and limiting sequence diversity. Our approach departs from this paradigm by generating binder sequences directly in a continuous latent space derived from a pretrained protein embedding model, without relying on predicted structures, thereby improving structural and sequence diversity. To encourage the model to capture binding-relevant features rather than memorizing known sequences, we perform latent-space exploration and diffusion-based sampling, enabling the generation of peptides beyond the limited distribution of known binders. This zero-shot generative strategy leverages the global protein embedding manifold as a semantic prior, allowing the model to propose novel peptide sequences in previously unseen regions of the protein space. We evaluate PepEDiff on TIGIT, a challenging target with a large, flat protein-protein interaction interface that lacks a druggable pocket. Despite its simplicity, our method outperforms state-of-the-art approaches across benchmark tests and in the TIGIT case study, demonstrating its potential as a general, structure-free framework for zero-shot peptide binder design. The code for this research is available at GitHub: https://github.com/LabJunBMI/PepEDiff-An-Peptide-binder-Embedding-Diffusion-Model
Native Parallel Reasoner: Reasoning in Parallelism via Self-Distilled Reinforcement Learning
Dec 19, 2025We introduce Native Parallel Reasoner (NPR), a teacher-free framework that enables Large Language Models (LLMs) to self-evolve genuine parallel reasoning capabilities. NPR transforms the model from sequential emulation to native parallel cognition through three key innovations: 1) a self-distilled progressive training paradigm that transitions from ``cold-start'' format discovery to strict topological constraints without external supervision; 2) a novel Parallel-Aware Policy Optimization (PAPO) algorithm that optimizes branching policies directly within the execution graph, allowing the model to learn adaptive decomposition via trial and error; and 3) a robust NPR Engine that refactors memory management and flow control of SGLang to enable stable, large-scale parallel RL training. Across eight reasoning benchmarks, NPR trained on Qwen3-4B achieves performance gains of up to 24.5% and inference speedups up to 4.6x. Unlike prior baselines that often fall back to autoregressive decoding, NPR demonstrates 100% genuine parallel execution, establishing a new standard for self-evolving, efficient, and scalable agentic reasoning.
Cross-Modal Unlearning via Influential Neuron Path Editing in Multimodal Large Language Models
Nov 10, 2025Multimodal Large Language Models (MLLMs) extend foundation models to real-world applications by integrating inputs such as text and vision. However, their broad knowledge capacity raises growing concerns about privacy leakage, toxicity mitigation, and intellectual property violations. Machine Unlearning (MU) offers a practical solution by selectively forgetting targeted knowledge while preserving overall model utility. When applied to MLLMs, existing neuron-editing-based MU approaches face two fundamental challenges: (1) forgetting becomes inconsistent across modalities because existing point-wise attribution methods fail to capture the structured, layer-by-layer information flow that connects different modalities; and (2) general knowledge performance declines when sensitive neurons that also support important reasoning paths are pruned, as this disrupts the model's ability to generalize. To alleviate these limitations, we propose a multimodal influential neuron path editor (MIP-Editor) for MU. Our approach introduces modality-specific attribution scores to identify influential neuron paths responsible for encoding forget-set knowledge and applies influential-path-aware neuron-editing via representation misdirection. This strategy also enables effective and coordinated forgetting across modalities while preserving the model's general capabilities. Experimental results demonstrate that MIP-Editor achieves a superior unlearning performance on multimodal tasks, with a maximum forgetting rate of 87.75% and up to 54.26% improvement in general knowledge retention. On textual tasks, MIP-Editor achieves up to 80.65% forgetting and preserves 77.9% of general performance. Codes are available at https://github.com/PreckLi/MIP-Editor.
Federated Unlearning in the Wild: Rethinking Fairness and Data Discrepancy
Oct 08, 2025Machine unlearning is critical for enforcing data deletion rights like the "right to be forgotten." As a decentralized paradigm, Federated Learning (FL) also requires unlearning, but realistic implementations face two major challenges. First, fairness in Federated Unlearning (FU) is often overlooked. Exact unlearning methods typically force all clients into costly retraining, even those uninvolved. Approximate approaches, using gradient ascent or distillation, make coarse interventions that can unfairly degrade performance for clients with only retained data. Second, most FU evaluations rely on synthetic data assumptions (IID/non-IID) that ignore real-world heterogeneity. These unrealistic benchmarks obscure the true impact of unlearning and limit the applicability of current methods. We first conduct a comprehensive benchmark of existing FU methods under realistic data heterogeneity and fairness conditions. We then propose a novel, fairness-aware FU approach, Federated Cross-Client-Constrains Unlearning (FedCCCU), to explicitly address both challenges. FedCCCU offers a practical and scalable solution for real-world FU. Experimental results show that existing methods perform poorly in realistic settings, while our approach consistently outperforms them.
Understanding and Leveraging the Expert Specialization of Context Faithfulness in Mixture-of-Experts LLMs
Aug 27, 2025
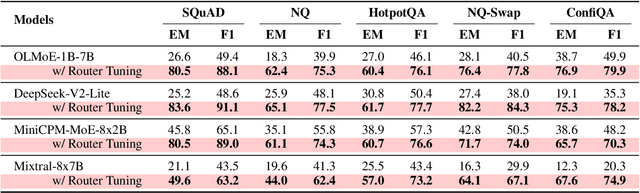
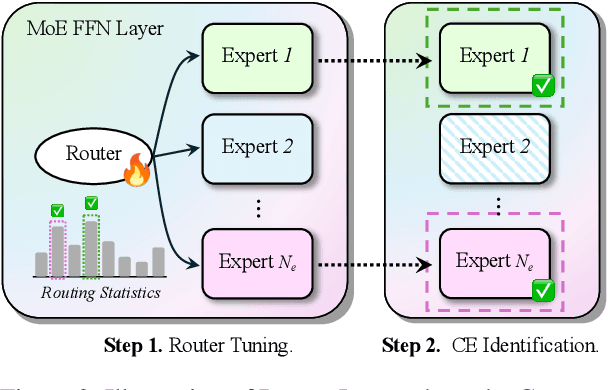
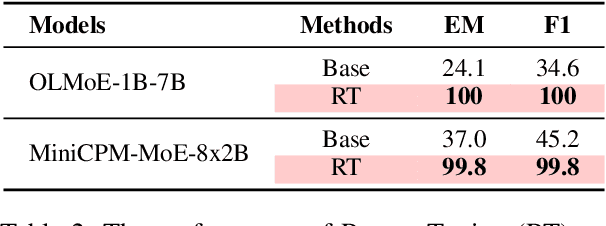
Context faithfulness is essential for reliable reasoning in context-dependent scenarios. However, large language models often struggle to ground their outputs in the provided context, resulting in irrelevant responses. Inspired by the emergent expert specialization observed in mixture-of-experts architectures, this work investigates whether certain experts exhibit specialization in context utilization, offering a potential pathway toward targeted optimization for improved context faithfulness. To explore this, we propose Router Lens, a method that accurately identifies context-faithful experts. Our analysis reveals that these experts progressively amplify attention to relevant contextual information, thereby enhancing context grounding. Building on this insight, we introduce Context-faithful Expert Fine-Tuning (CEFT), a lightweight optimization approach that selectively fine-tunes context-faithful experts. Experiments across a wide range of benchmarks and models demonstrate that CEFT matches or surpasses the performance of full fine-tuning while being significantly more efficient.
TongSearch-QR: Reinforced Query Reasoning for Retrieval
Jun 16, 2025



Traditional information retrieval (IR) methods excel at textual and semantic matching but struggle in reasoning-intensive retrieval tasks that require multi-hop inference or complex semantic understanding between queries and documents. One promising solution is to explicitly rewrite or augment queries using large language models (LLMs) to elicit reasoning-relevant content prior to retrieval. However, the widespread use of large-scale language models like GPT-4 or LLaMA3-70B remains impractical due to their high inference cost and limited deployability in real-world systems. In this work, we introduce TongSearch QR (Previously Known as "TongSearch Reasoner"), a family of small-scale language models for query reasoning and rewriting in reasoning-intensive retrieval. With a novel semi-rule-based reward function, we employ reinforcement learning approaches enabling smaller language models, e,g, Qwen2.5-7B-Instruct and Qwen2.5-1.5B-Instruct, to achieve query reasoning performance rivaling large-scale language models without their prohibitive inference costs. Experiment results on BRIGHT benchmark show that with BM25 as retrievers, both TongSearch QR-7B and TongSearch QR-1.5B models significantly outperform existing baselines, including prompt-based query reasoners and some latest dense retrievers trained for reasoning-intensive retrieval tasks, offering superior adaptability for real-world deployment.
FedMLAC: Mutual Learning Driven Heterogeneous Federated Audio Classification
Jun 11, 2025
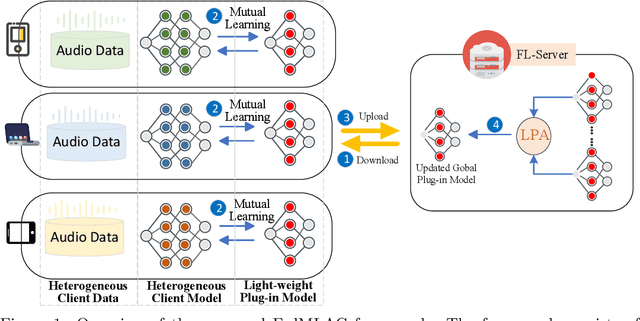

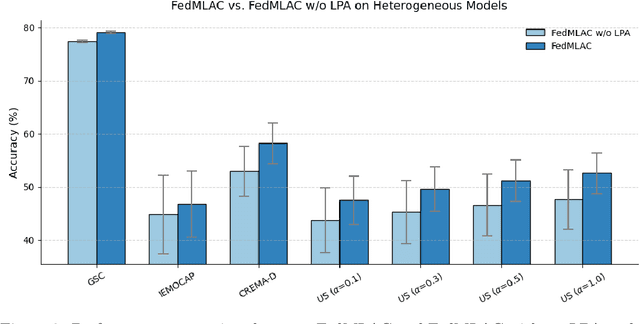
Federated Learning (FL) provides a privacy-preserving paradigm for training audio classification (AC) models across distributed clients without sharing raw data. However, Federated Audio Classification (FedAC) faces three critical challenges that substantially hinder performance: data heterogeneity, model heterogeneity, and data poisoning. While prior works have attempted to address these issues, they are typically treated independently, lacking a unified and robust solution suited to real-world federated audio scenarios. To bridge this gap, we propose FedMLAC, a unified mutual learning framework designed to simultaneously tackle these challenges in FedAC. Specifically, FedMLAC introduces a dual-model architecture on each client, comprising a personalized local AC model and a lightweight, globally shared Plug-in model. Through bidirectional knowledge distillation, the Plug-in model enables global knowledge transfer while adapting to client-specific data distributions, thus supporting both generalization and personalization. To further enhance robustness against corrupted audio data, we develop a Layer-wise Pruning Aggregation (LPA) strategy that filters unreliable Plug-in model updates based on parameter deviations during server-side aggregation. Extensive experiments on four diverse audio classification benchmarks, spanning both speech and non-speech tasks, demonstrate that FedMLAC consistently outperforms existing state-of-the-art methods in terms of classification accuracy and robustness to noisy data.
Selecting Demonstrations for Many-Shot In-Context Learning via Gradient Matching
Jun 05, 2025



In-Context Learning (ICL) empowers Large Language Models (LLMs) for rapid task adaptation without Fine-Tuning (FT), but its reliance on demonstration selection remains a critical challenge. While many-shot ICL shows promising performance through scaled demonstrations, the selection method for many-shot demonstrations remains limited to random selection in existing work. Since the conventional instance-level retrieval is not suitable for many-shot scenarios, we hypothesize that the data requirements for in-context learning and fine-tuning are analogous. To this end, we introduce a novel gradient matching approach that selects demonstrations by aligning fine-tuning gradients between the entire training set of the target task and the selected examples, so as to approach the learning effect on the entire training set within the selected examples. Through gradient matching on relatively small models, e.g., Qwen2.5-3B or Llama3-8B, our method consistently outperforms random selection on larger LLMs from 4-shot to 128-shot scenarios across 9 diverse datasets. For instance, it surpasses random selection by 4% on Qwen2.5-72B and Llama3-70B, and by around 2% on 5 closed-source LLMs. This work unlocks more reliable and effective many-shot ICL, paving the way for its broader application.
Disentangling Preference Representation and Text Generation for Efficient Individual Preference Alignment
Dec 30, 2024Aligning Large Language Models (LLMs) with general human preferences has been proved crucial in improving the interaction quality between LLMs and human. However, human values are inherently diverse among different individuals, making it insufficient to align LLMs solely with general preferences. To address this, personalizing LLMs according to individual feedback emerges as a promising solution. Nonetheless, this approach presents challenges in terms of the efficiency of alignment algorithms. In this work, we introduce a flexible paradigm for individual preference alignment. Our method fundamentally improves efficiency by disentangling preference representation from text generation in LLMs. We validate our approach across multiple text generation tasks and demonstrate that it can produce aligned quality as well as or better than PEFT-based methods, while reducing additional training time for each new individual preference by $80\%$ to $90\%$ in comparison with them.