 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeIncremental Transformer Neural Processes
Feb 21, 2026Neural Processes (NPs), and specifically Transformer Neural Processes (TNPs), have demonstrated remarkable performance across tasks ranging from spatiotemporal forecasting to tabular data modelling. However, many of these applications are inherently sequential, involving continuous data streams such as real-time sensor readings or database updates. In such settings, models should support cheap, incremental updates rather than recomputing internal representations from scratch for every new observation -- a capability existing TNP variants lack. Drawing inspiration from Large Language Models, we introduce the Incremental TNP (incTNP). By leveraging causal masking, Key-Value (KV) caching, and a data-efficient autoregressive training strategy, incTNP matches the predictive performance of standard TNPs while reducing the computational cost of updates from quadratic to linear time complexity. We empirically evaluate our model on a range of synthetic and real-world tasks, including tabular regression and temperature prediction. Our results show that, surprisingly, incTNP delivers performance comparable to -- or better than -- non-causal TNPs while unlocking orders-of-magnitude speedups for sequential inference. Finally, we assess the consistency of the model's updates -- by adapting a metric of ``implicit Bayesianness", we show that incTNP retains a prediction rule as implicitly Bayesian as standard non-causal TNPs, demonstrating that incTNP achieves the computational benefits of causal masking without sacrificing the consistency required for streaming inference.
Probabilistic Modelling is Sufficient for Causal Inference
Dec 29, 2025Causal inference is a key research area in machine learning, yet confusion reigns over the tools needed to tackle it. There are prevalent claims in the machine learning literature that you need a bespoke causal framework or notation to answer causal questions. In this paper, we want to make it clear that you \emph{can} answer any causal inference question within the realm of probabilistic modelling and inference, without causal-specific tools or notation. Through concrete examples, we demonstrate how causal questions can be tackled by writing down the probability of everything. Lastly, we reinterpret causal tools as emerging from standard probabilistic modelling and inference, elucidating their necessity and utility.
Completed Hyperparameter Transfer across Modules, Width, Depth, Batch and Duration
Dec 26, 2025Hyperparameter tuning can dramatically impact training stability and final performance of large-scale models. Recent works on neural network parameterisations, such as $μ$P, have enabled transfer of optimal global hyperparameters across model sizes. These works propose an empirical practice of search for optimal global base hyperparameters at a small model size, and transfer to a large size. We extend these works in two key ways. To handle scaling along most important scaling axes, we propose the Complete$^{(d)}$ Parameterisation that unifies scaling in width and depth -- using an adaptation of CompleteP -- as well as in batch-size and training duration. Secondly, with our parameterisation, we investigate per-module hyperparameter optimisation and transfer. We characterise the empirical challenges of navigating the high-dimensional hyperparameter landscape, and propose practical guidelines for tackling this optimisation problem. We demonstrate that, with the right parameterisation, hyperparameter transfer holds even in the per-module hyperparameter regime. Our study covers an extensive range of optimisation hyperparameters of modern models: learning rates, AdamW parameters, weight decay, initialisation scales, and residual block multipliers. Our experiments demonstrate significant training speed improvements in Large Language Models with the transferred per-module hyperparameters.
Wavelet-Induced Rotary Encodings: RoPE Meets Graphs
Sep 26, 2025We introduce WIRE: Wavelet-Induced Rotary Encodings. WIRE extends Rotary Position Encodings (RoPE), a popular algorithm in LLMs and ViTs, to graph-structured data. We demonstrate that WIRE is more general than RoPE, recovering the latter in the special case of grid graphs. WIRE also enjoys a host of desirable theoretical properties, including equivariance under node ordering permutation, compatibility with linear attention, and (under select assumptions) asymptotic dependence on graph resistive distance. We test WIRE on a range of synthetic and real-world tasks, including identifying monochromatic subgraphs, semantic segmentation of point clouds, and more standard graph benchmarks. We find it to be effective in settings where the underlying graph structure is important.
SKATE, a Scalable Tournament Eval: Weaker LLMs differentiate between stronger ones using verifiable challenges
Aug 08, 2025Evaluating the capabilities and risks of foundation models is paramount, yet current methods demand extensive domain expertise, hindering their scalability as these models rapidly evolve. We introduce SKATE: a novel evaluation framework in which large language models (LLMs) compete by generating and solving verifiable tasks for one another. Our core insight is to treat evaluation as a game: models act as both task-setters and solvers, incentivized to create questions which highlight their own strengths while exposing others' weaknesses. SKATE offers several key advantages, balancing scalability, open-endedness, and objectivity. It is fully automated, data-free, and scalable, requiring no human input or domain expertise. By using verifiable tasks rather than LLM judges, scoring is objective. Unlike domain-limited programmatically-generated benchmarks (e.g. chess-playing or spatial reasoning), having LLMs creatively pose challenges enables open-ended and scalable evaluation. As a proof of concept, we introduce LLM-set code-output-prediction (COP) challenges as a verifiable and extensible framework in which to test our approach. Using a TrueSkill-based ranking system, we evaluate six frontier LLMs and find that: (1) weaker models can reliably differentiate and score stronger ones, (2) LLM-based systems are capable of self-preferencing behavior, generating questions that align with their own capabilities, and (3) SKATE automatically surfaces fine-grained capability differences between models. Our findings are an important step towards general, scalable evaluation frameworks which can keep pace with LLM progress.
Distributional Training Data Attribution
Jun 15, 2025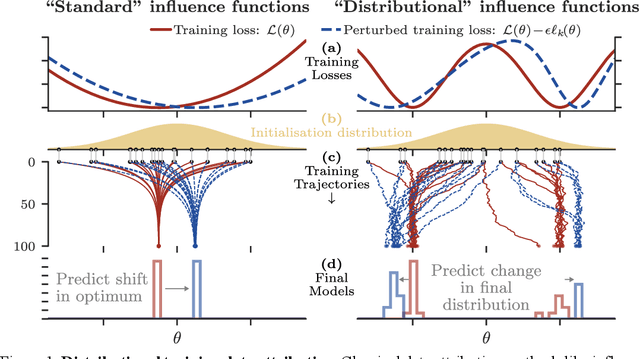
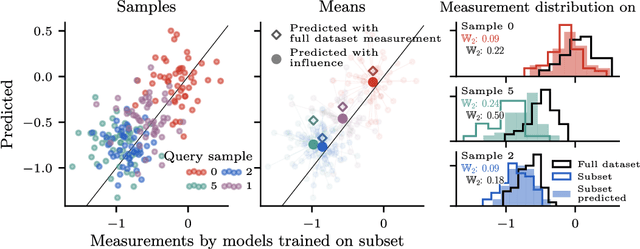
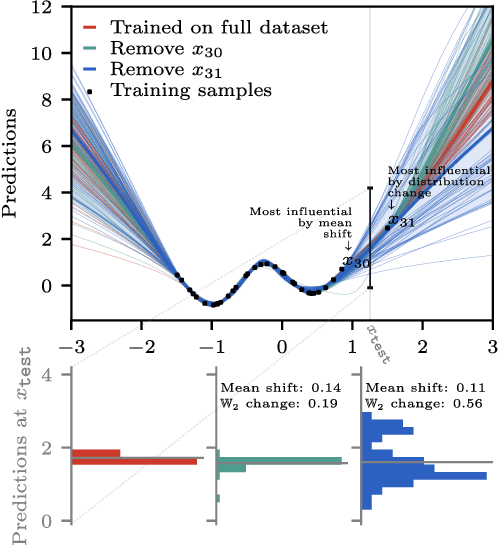
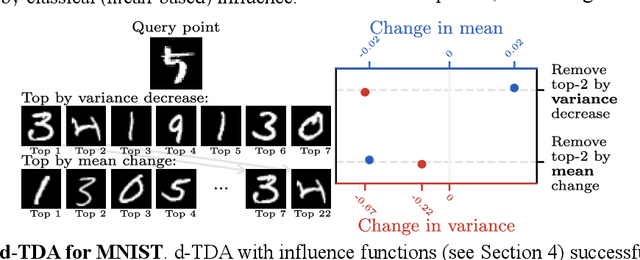
Randomness is an unavoidable part of training deep learning models, yet something that traditional training data attribution algorithms fail to rigorously account for. They ignore the fact that, due to stochasticity in the initialisation and batching, training on the same dataset can yield different models. In this paper, we address this shortcoming through introducing distributional training data attribution (d-TDA), the goal of which is to predict how the distribution of model outputs (over training runs) depends upon the dataset. We demonstrate the practical significance of d-TDA in experiments, e.g. by identifying training examples that drastically change the distribution of some target measurement without necessarily changing the mean. Intriguingly, we also find that influence functions (IFs), a popular but poorly-understood data attribution tool, emerge naturally from our distributional framework as the limit to unrolled differentiation; without requiring restrictive convexity assumptions. This provides a new mathematical motivation for their efficacy in deep learning, and helps to characterise their limitations.
Influence Functions for Scalable Data Attribution in Diffusion Models
Oct 17, 2024Diffusion models have led to significant advancements in generative modelling. Yet their widespread adoption poses challenges regarding data attribution and interpretability. In this paper, we aim to help address such challenges in diffusion models by developing an \textit{influence functions} framework. Influence function-based data attribution methods approximate how a model's output would have changed if some training data were removed. In supervised learning, this is usually used for predicting how the loss on a particular example would change. For diffusion models, we focus on predicting the change in the probability of generating a particular example via several proxy measurements. We show how to formulate influence functions for such quantities and how previously proposed methods can be interpreted as particular design choices in our framework. To ensure scalability of the Hessian computations in influence functions, we systematically develop K-FAC approximations based on generalised Gauss-Newton matrices specifically tailored to diffusion models. We recast previously proposed methods as specific design choices in our framework and show that our recommended method outperforms previous data attribution approaches on common evaluations, such as the Linear Data-modelling Score (LDS) or retraining without top influences, without the need for method-specific hyperparameter tuning.
Improving Linear System Solvers for Hyperparameter Optimisation in Iterative Gaussian Processes
May 28, 2024



Scaling hyperparameter optimisation to very large datasets remains an open problem in the Gaussian process community. This paper focuses on iterative methods, which use linear system solvers, like conjugate gradients, alternating projections or stochastic gradient descent, to construct an estimate of the marginal likelihood gradient. We discuss three key improvements which are applicable across solvers: (i) a pathwise gradient estimator, which reduces the required number of solver iterations and amortises the computational cost of making predictions, (ii) warm starting linear system solvers with the solution from the previous step, which leads to faster solver convergence at the cost of negligible bias, (iii) early stopping linear system solvers after a limited computational budget, which synergises with warm starting, allowing solver progress to accumulate over multiple marginal likelihood steps. These techniques provide speed-ups of up to $72\times$ when solving to tolerance, and decrease the average residual norm by up to $7\times$ when stopping early.
Warm Start Marginal Likelihood Optimisation for Iterative Gaussian Processes
May 28, 2024



Gaussian processes are a versatile probabilistic machine learning model whose effectiveness often depends on good hyperparameters, which are typically learned by maximising the marginal likelihood. In this work, we consider iterative methods, which use iterative linear system solvers to approximate marginal likelihood gradients up to a specified numerical precision, allowing a trade-off between compute time and accuracy of a solution. We introduce a three-level hierarchy of marginal likelihood optimisation for iterative Gaussian processes, and identify that the computational costs are dominated by solving sequential batches of large positive-definite systems of linear equations. We then propose to amortise computations by reusing solutions of linear system solvers as initialisations in the next step, providing a $\textit{warm start}$. Finally, we discuss the necessary conditions and quantify the consequences of warm starts and demonstrate their effectiveness on regression tasks, where warm starts achieve the same results as the conventional procedure while providing up to a $16 \times$ average speed-up among datasets.
Denoising Diffusion Probabilistic Models in Six Simple Steps
Feb 10, 2024Denoising Diffusion Probabilistic Models (DDPMs) are a very popular class of deep generative model that have been successfully applied to a diverse range of problems including image and video generation, protein and material synthesis, weather forecasting, and neural surrogates of partial differential equations. Despite their ubiquity it is hard to find an introduction to DDPMs which is simple, comprehensive, clean and clear. The compact explanations necessary in research papers are not able to elucidate all of the different design steps taken to formulate the DDPM and the rationale of the steps that are presented is often omitted to save space. Moreover, the expositions are typically presented from the variational lower bound perspective which is unnecessary and arguably harmful as it obfuscates why the method is working and suggests generalisations that do not perform well in practice. On the other hand, perspectives that take the continuous time-limit are beautiful and general, but they have a high barrier-to-entry as they require background knowledge of stochastic differential equations and probability flow. In this note, we distill down the formulation of the DDPM into six simple steps each of which comes with a clear rationale. We assume that the reader is familiar with fundamental topics in machine learning including basic probabilistic modelling, Gaussian distributions, maximum likelihood estimation, and deep learning.