 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtein Secondary Structure Prediction
Protein secondary-structure prediction is a vital task in bioinformatics, aiming to determine the arrangement of amino acids in proteins, including α-helices, β-sheets, and coils. By analyzing amino acid sequences, computational algorithms and machine learning techniques predict these structural elements. This knowledge is crucial for understanding protein function and interactions. While progress has been made, challenges remain, especially with non-local interactions and low sequence homology. Advancements in machine learning hold promise for improving prediction accuracy, furthering our understanding of protein biology.
Papers and Code
Towards Multiscale Graph-based Protein Learning with Geometric Secondary Structural Motifs
Jan 31, 2026Graph neural networks (GNNs) have emerged as powerful tools for learning protein structures by capturing spatial relationships at the residue level. However, existing GNN-based methods often face challenges in learning multiscale representations and modeling long-range dependencies efficiently. In this work, we propose an efficient multiscale graph-based learning framework tailored to proteins. Our proposed framework contains two crucial components: (1) It constructs a hierarchical graph representation comprising a collection of fine-grained subgraphs, each corresponding to a secondary structure motif (e.g., $α$-helices, $β$-strands, loops), and a single coarse-grained graph that connects these motifs based on their spatial arrangement and relative orientation. (2) It employs two GNNs for feature learning: the first operates within individual secondary motifs to capture local interactions, and the second models higher-level structural relationships across motifs. Our modular framework allows a flexible choice of GNN in each stage. Theoretically, we show that our hierarchical framework preserves the desired maximal expressiveness, ensuring no loss of critical structural information. Empirically, we demonstrate that integrating baseline GNNs into our multiscale framework remarkably improves prediction accuracy and reduces computational cost across various benchmarks.
Edge-aware GAT-based protein binding site prediction
Jan 05, 2026Accurate identification of protein binding sites is crucial for understanding biomolecular interaction mechanisms and for the rational design of drug targets. Traditional predictive methods often struggle to balance prediction accuracy with computational efficiency when capturing complex spatial conformations. To address this challenge, we propose an Edge-aware Graph Attention Network (Edge-aware GAT) model for the fine-grained prediction of binding sites across various biomolecules, including proteins, DNA/RNA, ions, ligands, and lipids. Our method constructs atom-level graphs and integrates multidimensional structural features, including geometric descriptors, DSSP-derived secondary structure, and relative solvent accessibility (RSA), to generate spatially aware embedding vectors. By incorporating interatomic distances and directional vectors as edge features within the attention mechanism, the model significantly enhances its representation capacity. On benchmark datasets, our model achieves an ROC-AUC of 0.93 for protein-protein binding site prediction, outperforming several state-of-the-art methods. The use of directional tensor propagation and residue-level attention pooling further improves both binding site localization and the capture of local structural details. Visualizations using PyMOL confirm the model's practical utility and interpretability. To facilitate community access and application, we have deployed a publicly accessible web server at http://119.45.201.89:5000/. In summary, our approach offers a novel and efficient solution that balances prediction accuracy, generalization, and interpretability for identifying functional sites in proteins.
Classifying Metamorphic versus Single-Fold Proteins with Statistical Learning and AlphaFold2
Dec 10, 2025The remarkable success of AlphaFold2 in providing accurate atomic-level prediction of protein structures from their amino acid sequence has transformed approaches to the protein folding problem. However, its core paradigm of mapping one sequence to one structure may only be appropriate for single-fold proteins with one stable conformation. Metamorphic proteins, which can adopt multiple distinct conformations, have conformational diversity that cannot be adequately modeled by AlphaFold2. Hence, classifying whether a given protein is metamorphic or single-fold remains a critical challenge for both laboratory experiments and computational methods. To address this challenge, we developed a novel classification framework by re-purposing AlphaFold2 to generate conformational ensembles via a multiple sequence alignment sampling method. From these ensembles, we extract a comprehensive set of features characterizing the conformational ensemble's modality and structural dispersion. A random forest classifier trained on a carefully curated benchmark dataset of known metamorphic and single-fold proteins achieves a mean AUC of 0.869 with cross-validation, demonstrating the effectiveness of our integrated approach. Furthermore, by applying our classifier to 600 randomly sampled proteins from the Protein Data Bank, we identified several potential metamorphic protein candidates -- including the 40S ribosomal protein S30, whose conformational change is crucial for its secondary function in antimicrobial defense. By combining AI-driven protein structure prediction with statistical learning, our work provides a powerful new approach for discovering metamorphic proteins and deepens our understanding of their role in their molecular function.
Ankh3: Multi-Task Pretraining with Sequence Denoising and Completion Enhances Protein Representations
May 26, 2025Protein language models (PLMs) have emerged as powerful tools to detect complex patterns of protein sequences. However, the capability of PLMs to fully capture information on protein sequences might be limited by focusing on single pre-training tasks. Although adding data modalities or supervised objectives can improve the performance of PLMs, pre-training often remains focused on denoising corrupted sequences. To push the boundaries of PLMs, our research investigated a multi-task pre-training strategy. We developed Ankh3, a model jointly optimized on two objectives: masked language modeling with multiple masking probabilities and protein sequence completion relying only on protein sequences as input. This multi-task pre-training demonstrated that PLMs can learn richer and more generalizable representations solely from protein sequences. The results demonstrated improved performance in downstream tasks, such as secondary structure prediction, fluorescence, GB1 fitness, and contact prediction. The integration of multiple tasks gave the model a more comprehensive understanding of protein properties, leading to more robust and accurate predictions.
A Comparative Review of RNA Language Models
May 14, 2025
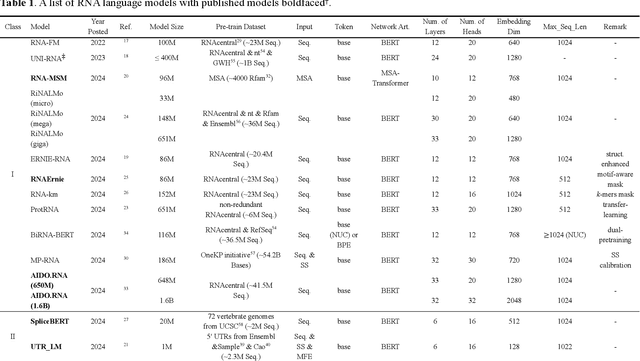

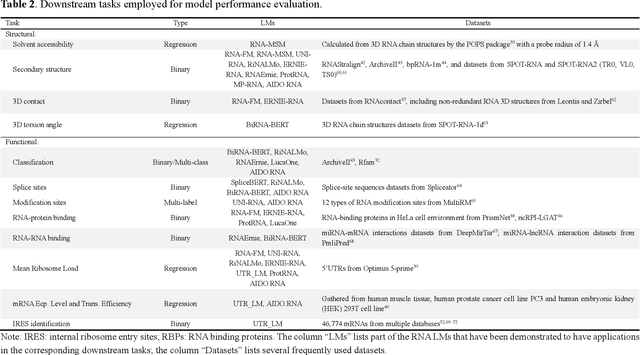
Given usefulness of protein language models (LMs) in structure and functional inference, RNA LMs have received increased attentions in the last few years. However, these RNA models are often not compared against the same standard. Here, we divided RNA LMs into three classes (pretrained on multiple RNA types (especially noncoding RNAs), specific-purpose RNAs, and LMs that unify RNA with DNA or proteins or both) and compared 13 RNA LMs along with 3 DNA and 1 protein LMs as controls in zero-shot prediction of RNA secondary structure and functional classification. Results shows that the models doing well on secondary structure prediction often perform worse in function classification or vice versa, suggesting that more balanced unsupervised training is needed.
ProtPainter: Draw or Drag Protein via Topology-guided Diffusion
Apr 19, 2025Recent advances in protein backbone generation have achieved promising results under structural, functional, or physical constraints. However, existing methods lack the flexibility for precise topology control, limiting navigation of the backbone space. We present ProtPainter, a diffusion-based approach for generating protein backbones conditioned on 3D curves. ProtPainter follows a two-stage process: curve-based sketching and sketch-guided backbone generation. For the first stage, we propose CurveEncoder, which predicts secondary structure annotations from a curve to parametrize sketch generation. For the second stage, the sketch guides the generative process in Denoising Diffusion Probabilistic Modeling (DDPM) to generate backbones. During this process, we further introduce a fusion scheduling scheme, Helix-Gating, to control the scaling factors. To evaluate, we propose the first benchmark for topology-conditioned protein generation, introducing Protein Restoration Task and a new metric, self-consistency Topology Fitness (scTF). Experiments demonstrate ProtPainter's ability to generate topology-fit (scTF > 0.8) and designable (scTM > 0.5) backbones, with drawing and dragging tasks showcasing its flexibility and versatility.
PLM-eXplain: Divide and Conquer the Protein Embedding Space
Apr 09, 2025Protein language models (PLMs) have revolutionised computational biology through their ability to generate powerful sequence representations for diverse prediction tasks. However, their black-box nature limits biological interpretation and translation to actionable insights. We present an explainable adapter layer - PLM-eXplain (PLM-X), that bridges this gap by factoring PLM embeddings into two components: an interpretable subspace based on established biochemical features, and a residual subspace that preserves the model's predictive power. Using embeddings from ESM2, our adapter incorporates well-established properties, including secondary structure and hydropathy while maintaining high performance. We demonstrate the effectiveness of our approach across three protein-level classification tasks: prediction of extracellular vesicle association, identification of transmembrane helices, and prediction of aggregation propensity. PLM-X enables biological interpretation of model decisions without sacrificing accuracy, offering a generalisable solution for enhancing PLM interpretability across various downstream applications. This work addresses a critical need in computational biology by providing a bridge between powerful deep learning models and actionable biological insights.
Customizing Spider Silk: Generative Models with Mechanical Property Conditioning for Protein Engineering
Apr 11, 2025The remarkable mechanical properties of spider silk, including its tensile strength and extensibility, are primarily governed by the repetitive regions of the proteins that constitute the fiber, the major ampullate spidroins (MaSps). However, establishing correlations between mechanical characteristics and repeat sequences is challenging due to the intricate sequence-structure-function relationships of MaSps and the limited availability of annotated datasets. In this study, we present a novel computational framework for designing MaSp repeat sequences with customizable mechanical properties. To achieve this, we developed a lightweight GPT-based generative model by distilling the pre-trained ProtGPT2 protein language model. The distilled model was subjected to multilevel fine-tuning using curated subsets of the Spider Silkome dataset. Specifically, we adapt the model for MaSp repeat generation using 6,000 MaSp repeat sequences and further refine it with 572 repeats associated with experimentally determined fiber-level mechanical properties. Our model generates biologically plausible MaSp repeat regions tailored to specific mechanical properties while also predicting those properties for given sequences. Validation includes sequence-level analysis, assessing physicochemical attributes and expected distribution of key motifs as well as secondary structure compositions. A correlation study using BLAST on the Spider Silkome dataset and a test set of MaSp repeats with known mechanical properties further confirmed the predictive accuracy of the model. This framework advances the rational design of spider silk-inspired biomaterials, offering a versatile tool for engineering protein sequences with tailored mechanical attributes.
Comprehensive benchmarking of large language models for RNA secondary structure prediction
Oct 21, 2024Inspired by the success of large language models (LLM) for DNA and proteins, several LLM for RNA have been developed recently. RNA-LLM uses large datasets of RNA sequences to learn, in a self-supervised way, how to represent each RNA base with a semantically rich numerical vector. This is done under the hypothesis that obtaining high-quality RNA representations can enhance data-costly downstream tasks. Among them, predicting the secondary structure is a fundamental task for uncovering RNA functional mechanisms. In this work we present a comprehensive experimental analysis of several pre-trained RNA-LLM, comparing them for the RNA secondary structure prediction task in an unified deep learning framework. The RNA-LLM were assessed with increasing generalization difficulty on benchmark datasets. Results showed that two LLM clearly outperform the other models, and revealed significant challenges for generalization in low-homology scenarios.
ProteinRPN: Towards Accurate Protein Function Prediction with Graph-Based Region Proposals
Sep 01, 2024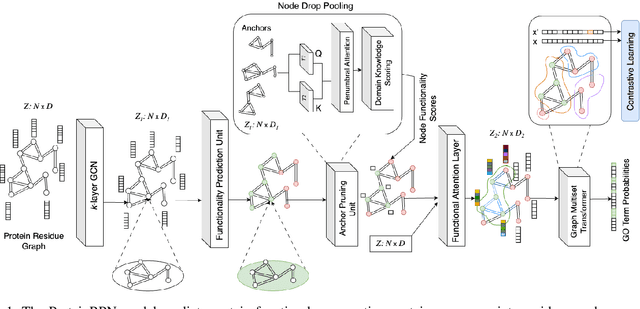
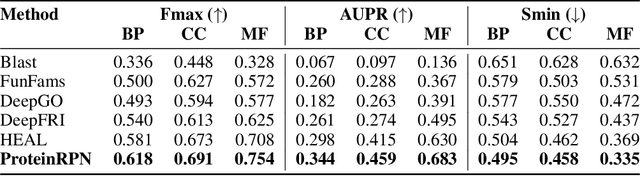
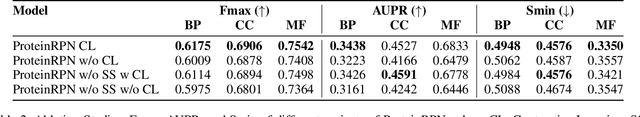
Protein function prediction is a crucial task in bioinformatics, with significant implications for understanding biological processes and disease mechanisms. While the relationship between sequence and function has been extensively explored, translating protein structure to function continues to present substantial challenges. Various models, particularly, CNN and graph-based deep learning approaches that integrate structural and functional data, have been proposed to address these challenges. However, these methods often fall short in elucidating the functional significance of key residues essential for protein functionality, as they predominantly adopt a retrospective perspective, leading to suboptimal performance. Inspired by region proposal networks in computer vision, we introduce the Protein Region Proposal Network (ProteinRPN) for accurate protein function prediction. Specifically, the region proposal module component of ProteinRPN identifies potential functional regions (anchors) which are refined through the hierarchy-aware node drop pooling layer favoring nodes with defined secondary structures and spatial proximity. The representations of the predicted functional nodes are enriched using attention mechanisms and subsequently fed into a Graph Multiset Transformer, which is trained with supervised contrastive (SupCon) and InfoNCE losses on perturbed protein structures. Our model demonstrates significant improvements in predicting Gene Ontology (GO) terms, effectively localizing functional residues within protein structures. The proposed framework provides a robust, scalable solution for protein function annotation, advancing the understanding of protein structure-function relationships in computational biology.