 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTowards a Science of Scaling Agent Systems
Dec 17, 2025Agents, language model-based systems that are capable of reasoning, planning, and acting are becoming the dominant paradigm for real-world AI applications. Despite this widespread adoption, the principles that determine their performance remain underexplored. We address this by deriving quantitative scaling principles for agent systems. We first formalize a definition for agentic evaluation and characterize scaling laws as the interplay between agent quantity, coordination structure, model capability, and task properties. We evaluate this across four benchmarks: Finance-Agent, BrowseComp-Plus, PlanCraft, and Workbench. With five canonical agent architectures (Single-Agent and four Multi-Agent Systems: Independent, Centralized, Decentralized, Hybrid), instantiated across three LLM families, we perform a controlled evaluation spanning 180 configurations. We derive a predictive model using coordination metrics, that achieves cross-validated R^2=0.524, enabling prediction on unseen task domains. We identify three effects: (1) a tool-coordination trade-off: under fixed computational budgets, tool-heavy tasks suffer disproportionately from multi-agent overhead. (2) a capability saturation: coordination yields diminishing or negative returns once single-agent baselines exceed ~45%. (3) topology-dependent error amplification: independent agents amplify errors 17.2x, while centralized coordination contains this to 4.4x. Centralized coordination improves performance by 80.8% on parallelizable tasks, while decentralized coordination excels on web navigation (+9.2% vs. +0.2%). Yet for sequential reasoning tasks, every multi-agent variants degraded performance by 39-70%. The framework predicts the optimal coordination strategy for 87% of held-out configurations. Out-of-sample validation on GPT-5.2, achieves MAE=0.071 and confirms four of five scaling principles generalize to unseen frontier models.
Completion $ eq$ Collaboration: Scaling Collaborative Effort with Agents
Oct 30, 2025Current evaluations of agents remain centered around one-shot task completion, failing to account for the inherently iterative and collaborative nature of many real-world problems, where human goals are often underspecified and evolve. We argue for a shift from building and assessing task completion agents to developing collaborative agents, assessed not only by the quality of their final outputs but by how well they engage with and enhance human effort throughout the problem-solving process. To support this shift, we introduce collaborative effort scaling, a framework that captures how an agent's utility grows with increasing user involvement. Through case studies and simulated evaluations, we show that state-of-the-art agents often underperform in multi-turn, real-world scenarios, revealing a missing ingredient in agent design: the ability to sustain engagement and scaffold user understanding. Collaborative effort scaling offers a lens for diagnosing agent behavior and guiding development toward more effective interactions.
RADAR: Benchmarking Language Models on Imperfect Tabular Data
Jun 09, 2025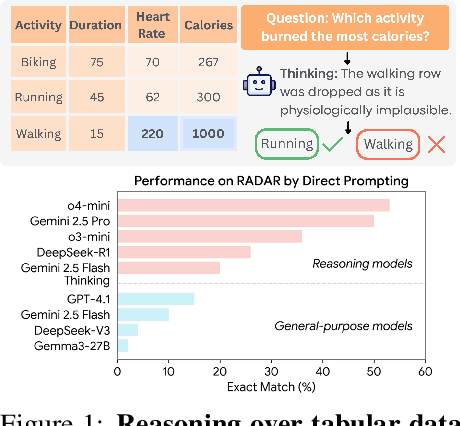
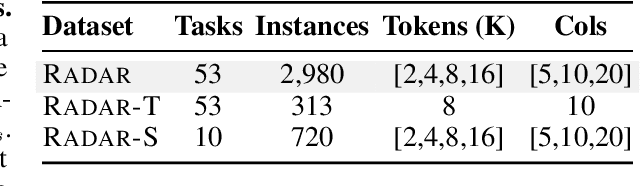
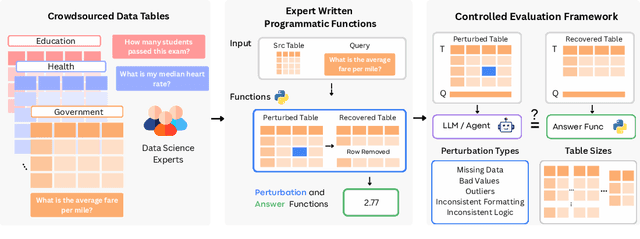
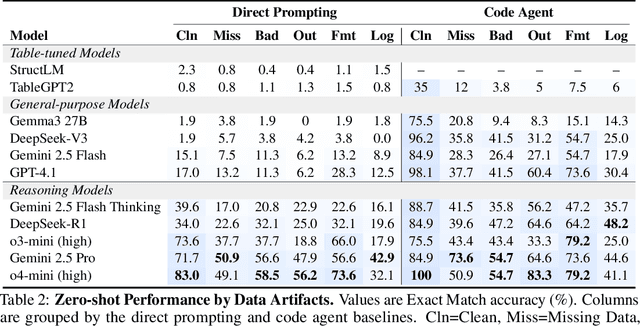
Language models (LMs) are increasingly being deployed to perform autonomous data analyses. However, their data awareness -- the ability to recognize, reason over, and appropriately handle data artifacts such as missing values, outliers, and logical inconsistencies -- remains underexplored. These artifacts are especially common in real-world tabular data and, if mishandled, can significantly compromise the validity of analytical conclusions. To address this gap, we present RADAR, a benchmark for systematically evaluating data-aware reasoning on tabular data. We develop a framework to simulate data artifacts via programmatic perturbations to enable targeted evaluation of model behavior. RADAR comprises 2980 table query pairs, grounded in real-world data spanning 9 domains and 5 data artifact types. In addition to evaluating artifact handling, RADAR systematically varies table size to study how reasoning performance holds when increasing table size. Our evaluation reveals that, despite decent performance on tables without data artifacts, frontier models degrade significantly when data artifacts are introduced, exposing critical gaps in their capacity for robust, data-aware analysis. Designed to be flexible and extensible, RADAR supports diverse perturbation types and controllable table sizes, offering a valuable resource for advancing tabular reasoning.
LSM-2: Learning from Incomplete Wearable Sensor Data
Jun 05, 2025



Foundation models, a cornerstone of recent advancements in machine learning, have predominantly thrived on complete and well-structured data. Wearable sensor data frequently suffers from significant missingness, posing a substantial challenge for self-supervised learning (SSL) models that typically assume complete data inputs. This paper introduces the second generation of Large Sensor Model (LSM-2) with Adaptive and Inherited Masking (AIM), a novel SSL approach that learns robust representations directly from incomplete data without requiring explicit imputation. AIM's core novelty lies in its use of learnable mask tokens to model both existing ("inherited") and artificially introduced missingness, enabling it to robustly handle fragmented real-world data during inference. Pre-trained on an extensive dataset of 40M hours of day-long multimodal sensor data, our LSM-2 with AIM achieves the best performance across a diverse range of tasks, including classification, regression and generative modeling. Furthermore, LSM-2 with AIM exhibits superior scaling performance, and critically, maintains high performance even under targeted missingness scenarios, reflecting clinically coherent patterns, such as the diagnostic value of nighttime biosignals for hypertension prediction. This makes AIM a more reliable choice for real-world wearable data applications.
BLADE: Benchmarking Language Model Agents for Data-Driven Science
Aug 20, 2024



Data-driven scientific discovery requires the iterative integration of scientific domain knowledge, statistical expertise, and an understanding of data semantics to make nuanced analytical decisions, e.g., about which variables, transformations, and statistical models to consider. LM-based agents equipped with planning, memory, and code execution capabilities have the potential to support data-driven science. However, evaluating agents on such open-ended tasks is challenging due to multiple valid approaches, partially correct steps, and different ways to express the same decisions. To address these challenges, we present BLADE, a benchmark to automatically evaluate agents' multifaceted approaches to open-ended research questions. BLADE consists of 12 datasets and research questions drawn from existing scientific literature, with ground truth collected from independent analyses by expert data scientists and researchers. To automatically evaluate agent responses, we developed corresponding computational methods to match different representations of analyses to this ground truth. Though language models possess considerable world knowledge, our evaluation shows that they are often limited to basic analyses. However, agents capable of interacting with the underlying data demonstrate improved, but still non-optimal, diversity in their analytical decision making. Our work enables the evaluation of agents for data-driven science and provides researchers deeper insights into agents' analysis approaches.
Bi-Level Graph Neural Networks for Drug-Drug Interaction Prediction
Jun 11, 2020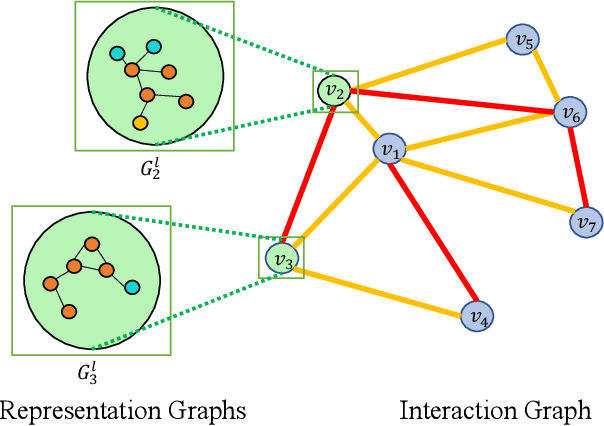
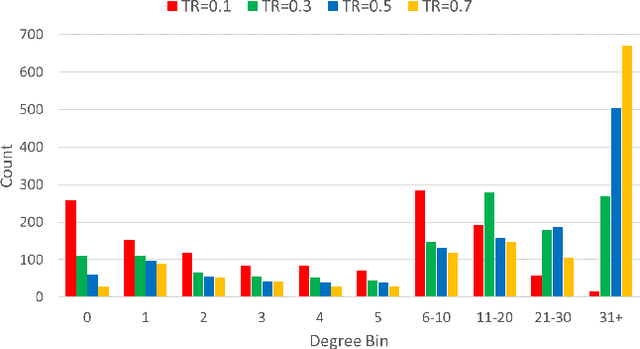
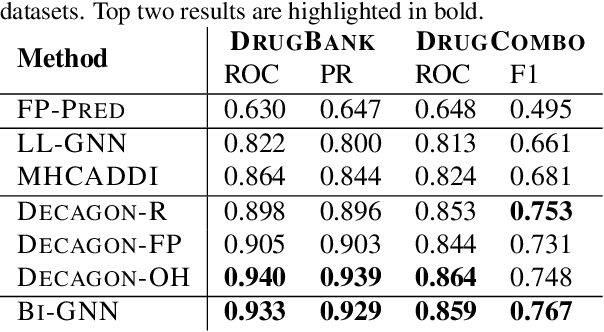
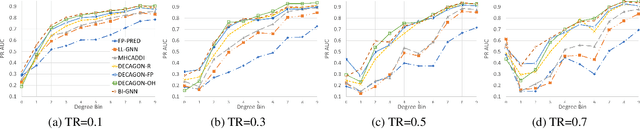
We introduce Bi-GNN for modeling biological link prediction tasks such as drug-drug interaction (DDI) and protein-protein interaction (PPI). Taking drug-drug interaction as an example, existing methods using machine learning either only utilize the link structure between drugs without using the graph representation of each drug molecule, or only leverage the individual drug compound structures without using graph structure for the higher-level DDI graph. The key idea of our method is to fundamentally view the data as a bi-level graph, where the highest level graph represents the interaction between biological entities (interaction graph), and each biological entity itself is further expanded to its intrinsic graph representation (representation graphs), where the graph is either flat like a drug compound or hierarchical like a protein with amino acid level graph, secondary structure, tertiary structure, etc. Our model not only allows the usage of information from both the high-level interaction graph and the low-level representation graphs, but also offers a baseline for future research opportunities to address the bi-level nature of the data.
Fast Detection of Maximum Common Subgraph via Deep Q-Learning
Feb 20, 2020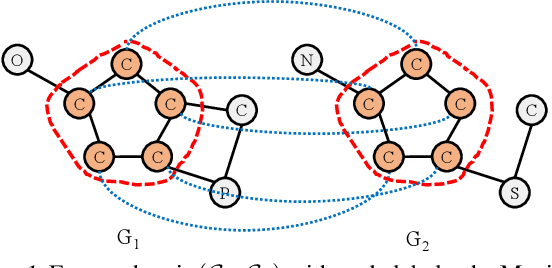

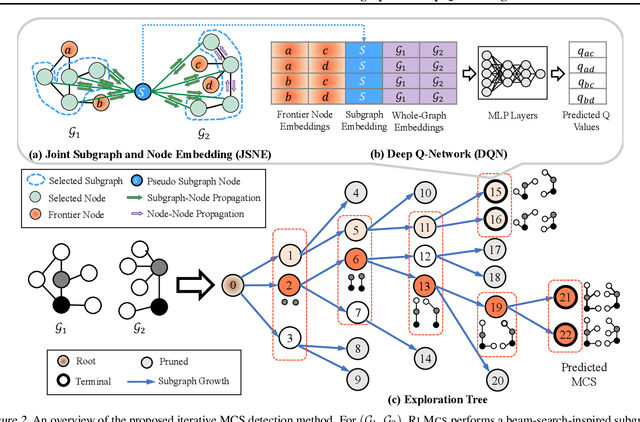
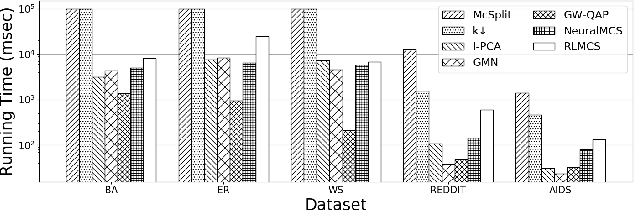
Detecting the Maximum Common Subgraph (MCS) between two input graphs is fundamental for applications in biomedical analysis, malware detection, cloud computing, etc. This is especially important in the task of drug design, where the successful extraction of common substructures in compounds can reduce the number of experiments needed to be conducted by humans. However, MCS computation is NP-hard, and state-of-the-art exact MCS solvers do not have worst-case time complexity guarantee and cannot handle large graphs in practice. Designing learning based models to find the MCS between two graphs in an approximate yet accurate way while utilizing as few labeled MCS instances as possible remains to be a challenging task. Here we propose RLMCS, a Graph Neural Network based model for MCS detection through reinforcement learning. Our model uses an exploration tree to extract subgraphs in two graphs one node pair at a time, and is trained to optimize subgraph extraction rewards via Deep Q-Networks. A novel graph embedding method is proposed to generate state representations for nodes and extracted subgraphs jointly at each step. Experiments on real graph datasets demonstrate that our model performs favorably to exact MCS solvers and supervised neural graph matching network models in terms of accuracy and efficiency.
Unsupervised Inductive Whole-Graph Embedding by Preserving Graph Proximity
Apr 01, 2019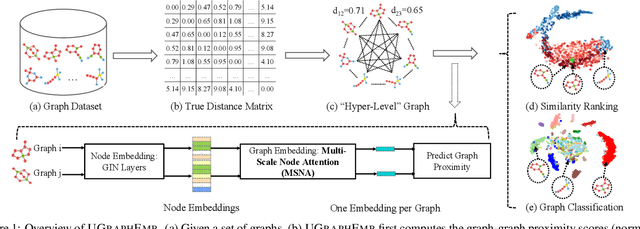



We introduce a novel approach to graph-level representation learning, which is to embed an entire graph into a vector space where the embeddings of two graphs preserve their graph-graph proximity. Our approach, UGRAPHEMB, is a general framework that provides a novel means to performing graph-level embedding in a completely unsupervised and inductive manner. The learned neural network can be considered as a function that receives any graph as input, either seen or unseen in the training set, and transforms it into an embedding. A novel graph-level embedding generation mechanism called Multi-Scale Node Attention (MSNA), is proposed. Experiments on five real graph datasets show that UGRAPHEMB achieves competitive accuracy in the tasks of graph classification, similarity ranking, and graph visualization.