 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGland segmentation via dual encoders and boundary-enhanced attention
Jan 29, 2024



Accurate and automated gland segmentation on pathological images can assist pathologists in diagnosing the malignancy of colorectal adenocarcinoma. However, due to various gland shapes, severe deformation of malignant glands, and overlapping adhesions between glands. Gland segmentation has always been very challenging. To address these problems, we propose a DEA model. This model consists of two branches: the backbone encoding and decoding network and the local semantic extraction network. The backbone encoding and decoding network extracts advanced Semantic features, uses the proposed feature decoder to restore feature space information, and then enhances the boundary features of the gland through boundary enhancement attention. The local semantic extraction network uses the pre-trained DeepLabv3+ as a Local semantic-guided encoder to realize the extraction of edge features. Experimental results on two public datasets, GlaS and CRAG, confirm that the performance of our method is better than other gland segmentation methods.
A Novel Dataset and a Deep Learning Method for Mitosis Nuclei Segmentation and Classification
Dec 27, 2022
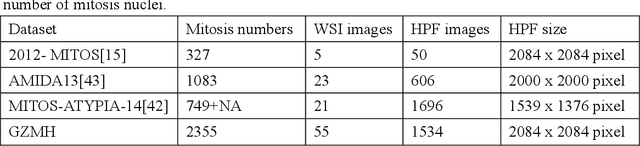
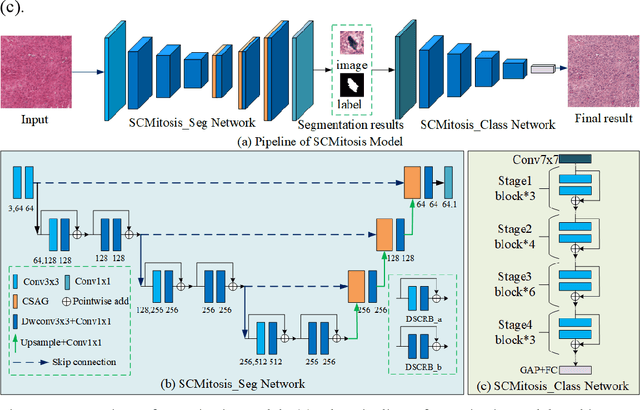
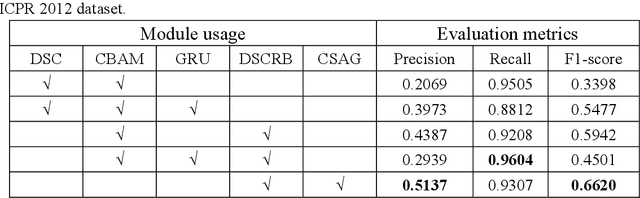
Mitosis nuclei count is one of the important indicators for the pathological diagnosis of breast cancer. The manual annotation needs experienced pathologists, which is very time-consuming and inefficient. With the development of deep learning methods, some models with good performance have emerged, but the generalization ability should be further strengthened. In this paper, we propose a two-stage mitosis segmentation and classification method, named SCMitosis. Firstly, the segmentation performance with a high recall rate is achieved by the proposed depthwise separable convolution residual block and channel-spatial attention gate. Then, a classification network is cascaded to further improve the detection performance of mitosis nuclei. The proposed model is verified on the ICPR 2012 dataset, and the highest F-score value of 0.8687 is obtained compared with the current state-of-the-art algorithms. In addition, the model also achieves good performance on GZMH dataset, which is prepared by our group and will be firstly released with the publication of this paper. The code will be available at: https://github.com/antifen/mitosis-nuclei-segmentation.
Binary Representation via Jointly Personalized Sparse Hashing
Aug 31, 2022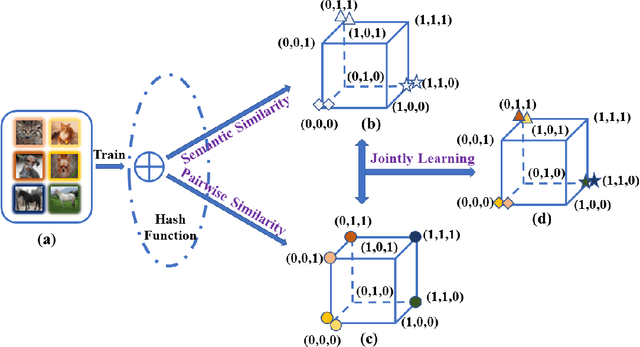
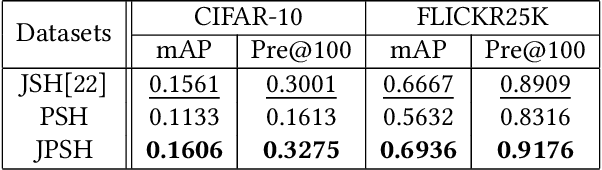
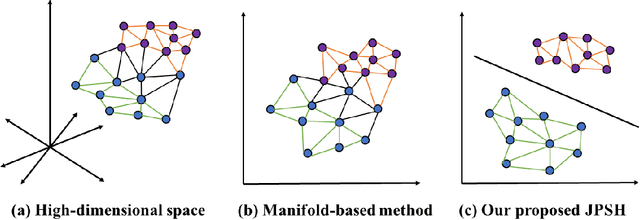
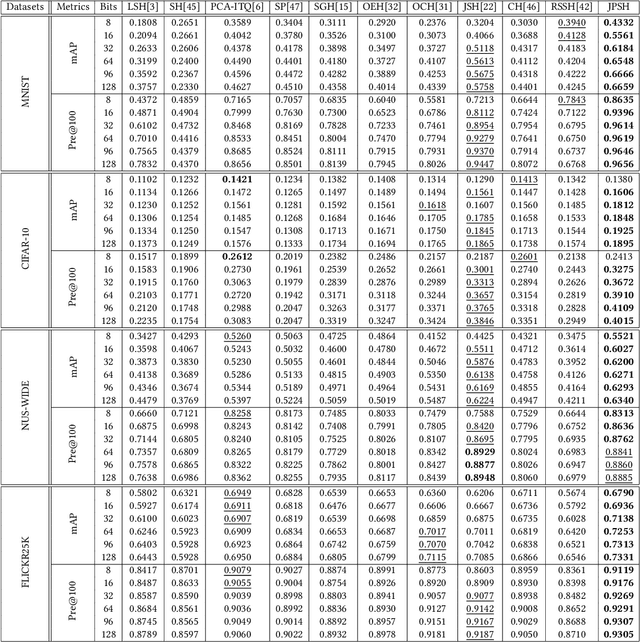
Unsupervised hashing has attracted much attention for binary representation learning due to the requirement of economical storage and efficiency of binary codes. It aims to encode high-dimensional features in the Hamming space with similarity preservation between instances. However, most existing methods learn hash functions in manifold-based approaches. Those methods capture the local geometric structures (i.e., pairwise relationships) of data, and lack satisfactory performance in dealing with real-world scenarios that produce similar features (e.g. color and shape) with different semantic information. To address this challenge, in this work, we propose an effective unsupervised method, namely Jointly Personalized Sparse Hashing (JPSH), for binary representation learning. To be specific, firstly, we propose a novel personalized hashing module, i.e., Personalized Sparse Hashing (PSH). Different personalized subspaces are constructed to reflect category-specific attributes for different clusters, adaptively mapping instances within the same cluster to the same Hamming space. In addition, we deploy sparse constraints for different personalized subspaces to select important features. We also collect the strengths of the other clusters to build the PSH module with avoiding over-fitting. Then, to simultaneously preserve semantic and pairwise similarities in our JPSH, we incorporate the PSH and manifold-based hash learning into the seamless formulation. As such, JPSH not only distinguishes the instances from different clusters, but also preserves local neighborhood structures within the cluster. Finally, an alternating optimization algorithm is adopted to iteratively capture analytical solutions of the JPSH model. Extensive experiments on four benchmark datasets verify that the JPSH outperforms several hashing algorithms on the similarity search task.
WSSS4LUAD: Grand Challenge on Weakly-supervised Tissue Semantic Segmentation for Lung Adenocarcinoma
Apr 14, 2022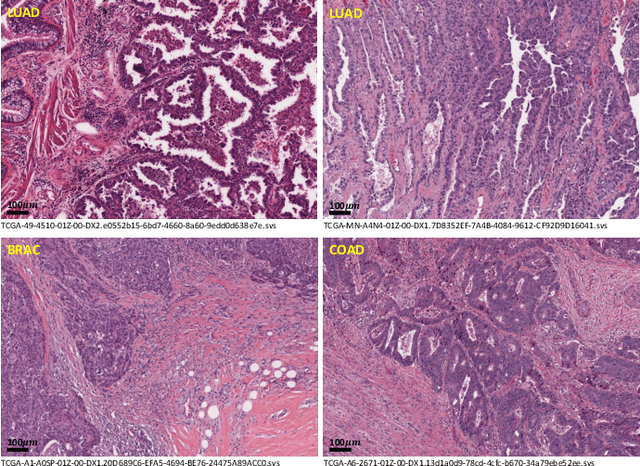
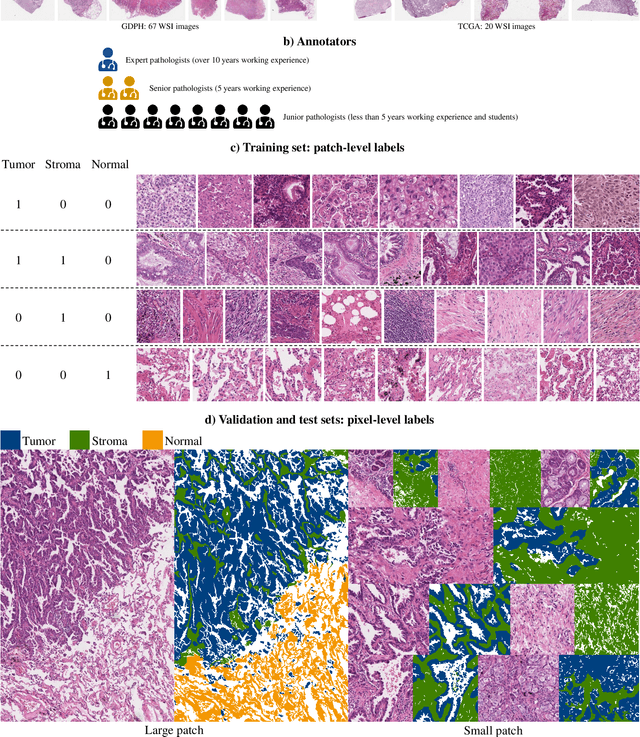
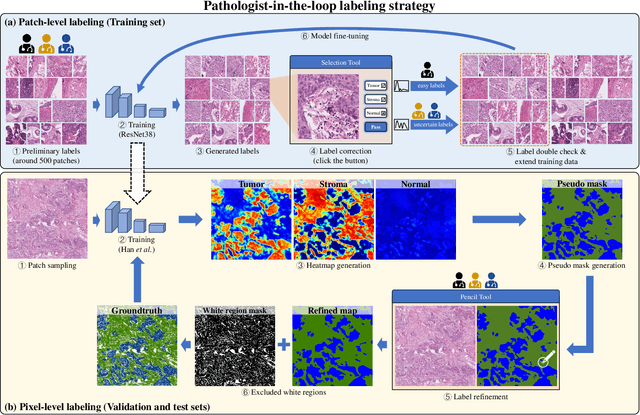
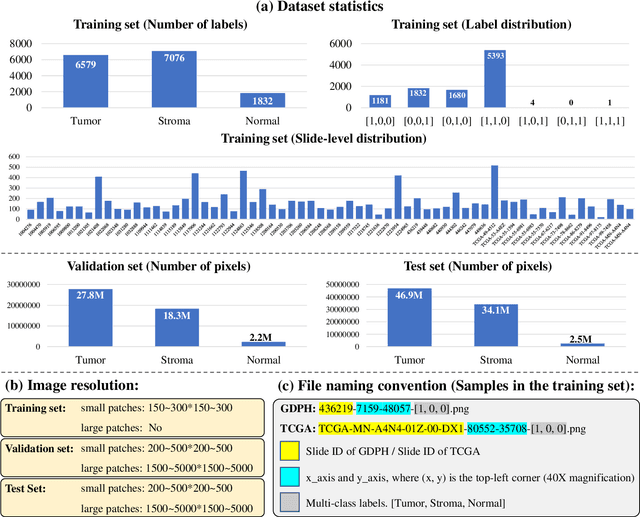
Lung cancer is the leading cause of cancer death worldwide, and adenocarcinoma (LUAD) is the most common subtype. Exploiting the potential value of the histopathology images can promote precision medicine in oncology. Tissue segmentation is the basic upstream task of histopathology image analysis. Existing deep learning models have achieved superior segmentation performance but require sufficient pixel-level annotations, which is time-consuming and expensive. To enrich the label resources of LUAD and to alleviate the annotation efforts, we organize this challenge WSSS4LUAD to call for the outstanding weakly-supervised semantic segmentation (WSSS) techniques for histopathology images of LUAD. Participants have to design the algorithm to segment tumor epithelial, tumor-associated stroma and normal tissue with only patch-level labels. This challenge includes 10,091 patch-level annotations (the training set) and over 130 million labeled pixels (the validation and test sets), from 87 WSIs (67 from GDPH, 20 from TCGA). All the labels were generated by a pathologist-in-the-loop pipeline with the help of AI models and checked by the label review board. Among 532 registrations, 28 teams submitted the results in the test phase with over 1,000 submissions. Finally, the first place team achieved mIoU of 0.8413 (tumor: 0.8389, stroma: 0.7931, normal: 0.8919). According to the technical reports of the top-tier teams, CAM is still the most popular approach in WSSS. Cutmix data augmentation has been widely adopted to generate more reliable samples. With the success of this challenge, we believe that WSSS approaches with patch-level annotations can be a complement to the traditional pixel annotations while reducing the annotation efforts. The entire dataset has been released to encourage more researches on computational pathology in LUAD and more novel WSSS techniques.
A Standardized Pipeline for Colon Nuclei Identification and Counting Challenge
Mar 20, 2022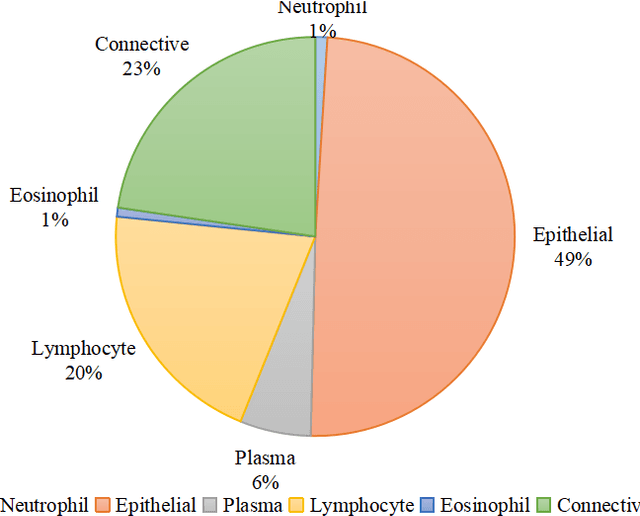
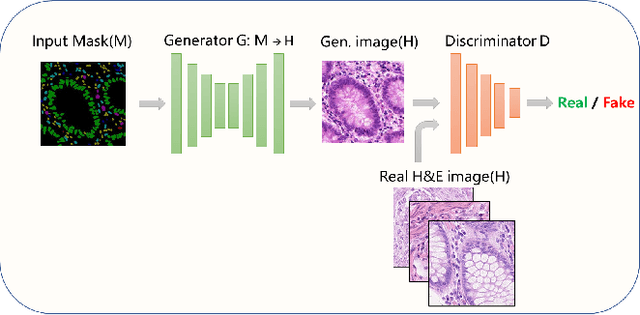
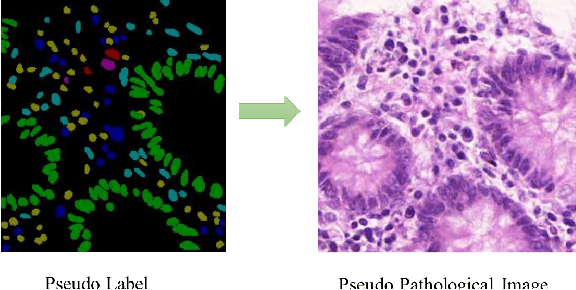
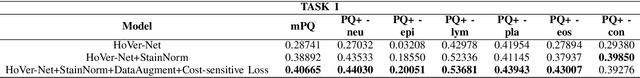
Nuclear segmentation and classification is an essential step for computational pathology. TIA lab from Warwick University organized a nuclear segmentation and classification challenge (CoNIC) for H&E stained histopathology images in colorectal cancer with two highly correlated tasks, nuclei segmentation and classification task and cellular composition task. There are a few obstacles we have to address in this challenge, 1) limited training samples, 2) color variation, 3) imbalanced annotations, 4) similar morphological appearance among classes. To deal with these challenges, we proposed a standardized pipeline for nuclear segmentation and classification by integrating several pluggable components. First, we built a GAN-based model to automatically generate pseudo images for data augmentation. Then we trained a self-supervised stain normalization model to solve the color variation problem. Next we constructed a baseline model HoVer-Net with cost-sensitive loss to encourage the model pay more attention on the minority classes. According to the results of the leaderboard, our proposed pipeline achieves 0.40665 mPQ+ (Rank 49th) and 0.62199 r2 (Rank 10th) in the preliminary test phase.
AIM 2020 Challenge on Efficient Super-Resolution: Methods and Results
Sep 15, 2020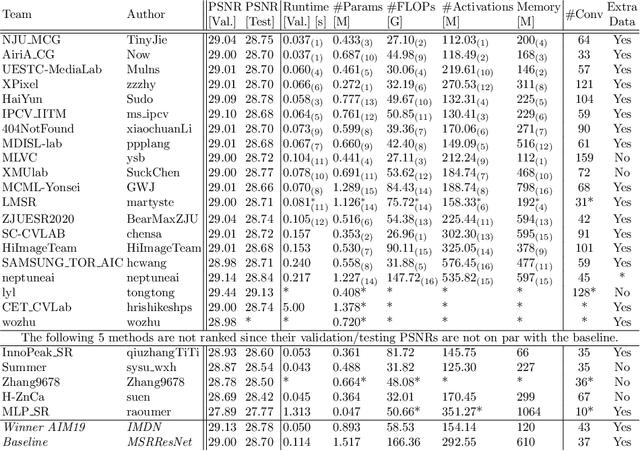
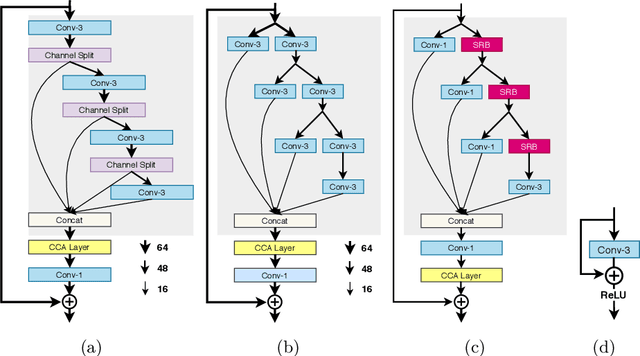
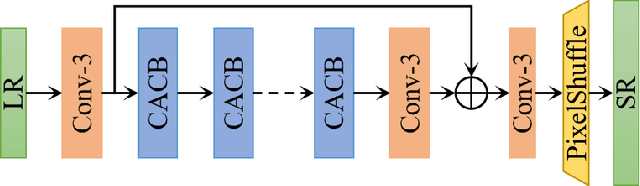
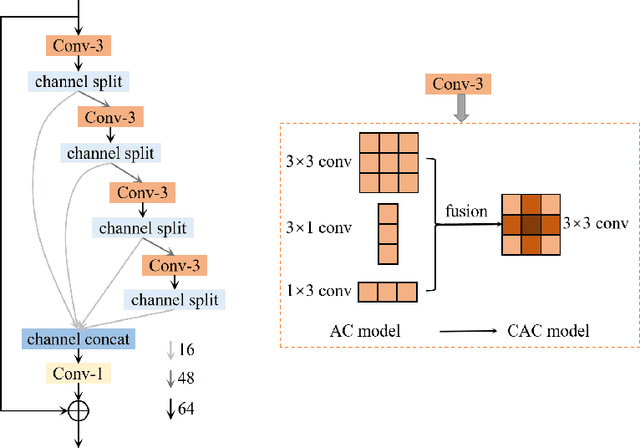
This paper reviews the AIM 2020 challenge on efficient single image super-resolution with focus on the proposed solutions and results. The challenge task was to super-resolve an input image with a magnification factor x4 based on a set of prior examples of low and corresponding high resolution images. The goal is to devise a network that reduces one or several aspects such as runtime, parameter count, FLOPs, activations, and memory consumption while at least maintaining PSNR of MSRResNet. The track had 150 registered participants, and 25 teams submitted the final results. They gauge the state-of-the-art in efficient single image super-resolution.