 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEvaluating Character Understanding of Large Language Models via Character Profiling from Fictional Works
Apr 19, 2024



Large language models (LLMs) have demonstrated impressive performance and spurred numerous AI applications, in which role-playing agents (RPAs) are particularly popular, especially for fictional characters. The prerequisite for these RPAs lies in the capability of LLMs to understand characters from fictional works. Previous efforts have evaluated this capability via basic classification tasks or characteristic imitation, failing to capture the nuanced character understanding with LLMs. In this paper, we propose evaluating LLMs' character understanding capability via the character profiling task, i.e., summarizing character profiles from corresponding materials, a widely adopted yet understudied practice for RPA development. Specifically, we construct the CroSS dataset from literature experts and assess the generated profiles by comparing ground truth references and their applicability in downstream tasks. Our experiments, which cover various summarization methods and LLMs, have yielded promising results. These results strongly validate the character understanding capability of LLMs. We believe our constructed resource will promote further research in this field. Resources are available at https://github.com/Joanna0123/character_profiling.
A Comprehensive Capability Analysis of GPT-3 and GPT-3.5 Series Models
Mar 18, 2023
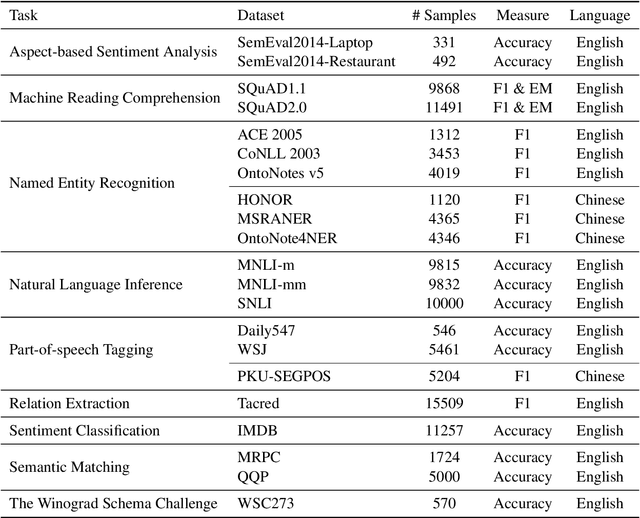
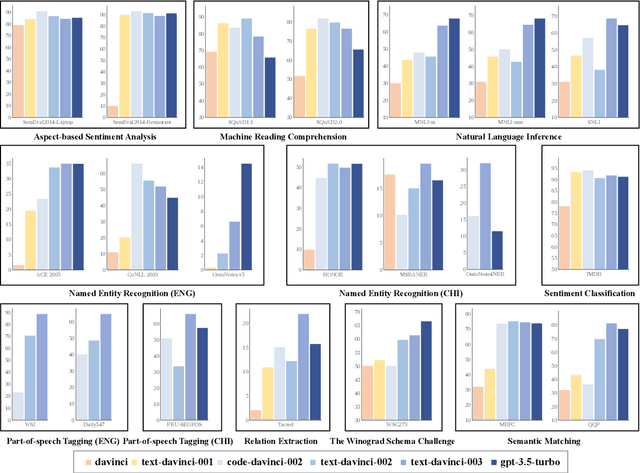
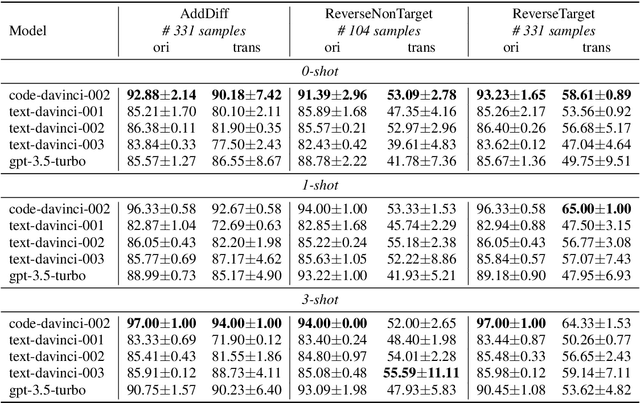
GPT series models, such as GPT-3, CodeX, InstructGPT, ChatGPT, and so on, have gained considerable attention due to their exceptional natural language processing capabilities. However, despite the abundance of research on the difference in capabilities between GPT series models and fine-tuned models, there has been limited attention given to the evolution of GPT series models' capabilities over time. To conduct a comprehensive analysis of the capabilities of GPT series models, we select six representative models, comprising two GPT-3 series models (i.e., davinci and text-davinci-001) and four GPT-3.5 series models (i.e., code-davinci-002, text-davinci-002, text-davinci-003, and gpt-3.5-turbo). We evaluate their performance on nine natural language understanding (NLU) tasks using 21 datasets. In particular, we compare the performance and robustness of different models for each task under zero-shot and few-shot scenarios. Our extensive experiments reveal that the overall ability of GPT series models on NLU tasks does not increase gradually as the models evolve, especially with the introduction of the RLHF training strategy. While this strategy enhances the models' ability to generate human-like responses, it also compromises their ability to solve some tasks. Furthermore, our findings indicate that there is still room for improvement in areas such as model robustness.
Gene-SGAN: a method for discovering disease subtypes with imaging and genetic signatures via multi-view weakly-supervised deep clustering
Jan 25, 2023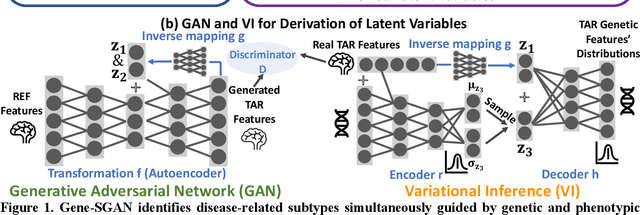
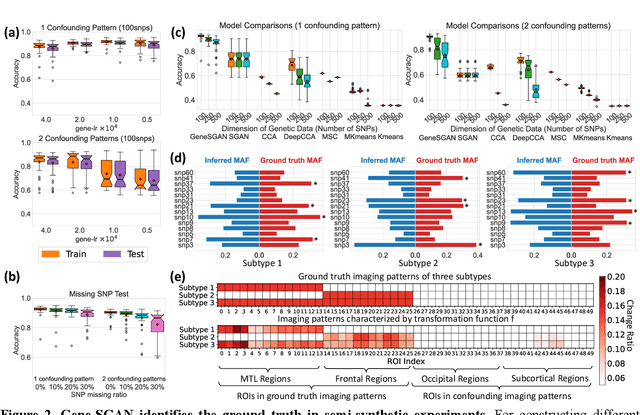
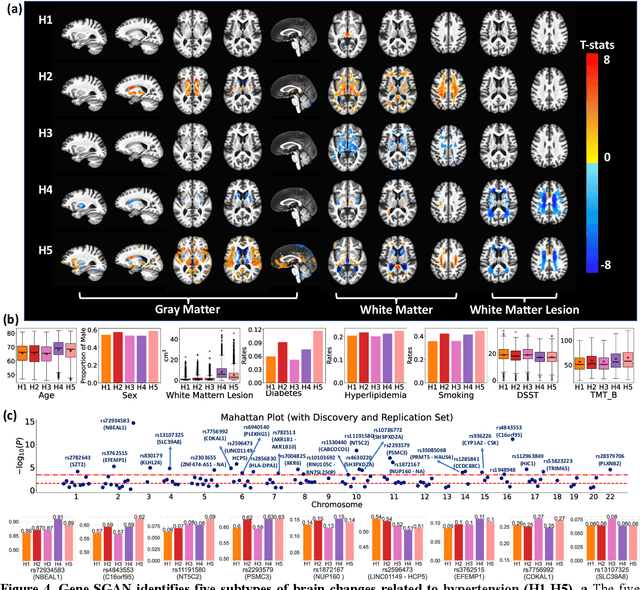
Disease heterogeneity has been a critical challenge for precision diagnosis and treatment, especially in neurologic and neuropsychiatric diseases. Many diseases can display multiple distinct brain phenotypes across individuals, potentially reflecting disease subtypes that can be captured using MRI and machine learning methods. However, biological interpretability and treatment relevance are limited if the derived subtypes are not associated with genetic drivers or susceptibility factors. Herein, we describe Gene-SGAN - a multi-view, weakly-supervised deep clustering method - which dissects disease heterogeneity by jointly considering phenotypic and genetic data, thereby conferring genetic correlations to the disease subtypes and associated endophenotypic signatures. We first validate the generalizability, interpretability, and robustness of Gene-SGAN in semi-synthetic experiments. We then demonstrate its application to real multi-site datasets from 28,858 individuals, deriving subtypes of Alzheimer's disease and brain endophenotypes associated with hypertension, from MRI and SNP data. Derived brain phenotypes displayed significant differences in neuroanatomical patterns, genetic determinants, biological and clinical biomarkers, indicating potentially distinct underlying neuropathologic processes, genetic drivers, and susceptibility factors. Overall, Gene-SGAN is broadly applicable to disease subtyping and endophenotype discovery, and is herein tested on disease-related, genetically-driven neuroimaging phenotypes.
STREAMLINE: A Simple, Transparent, End-To-End Automated Machine Learning Pipeline Facilitating Data Analysis and Algorithm Comparison
Jun 23, 2022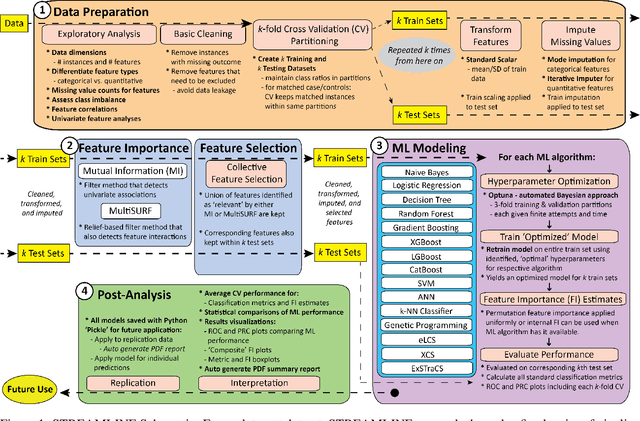
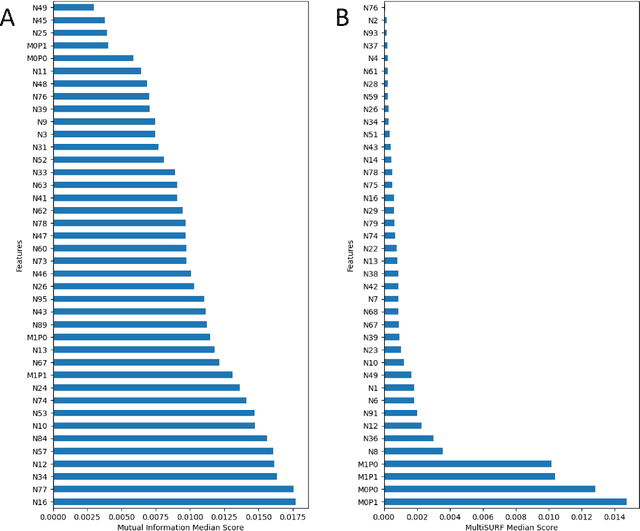
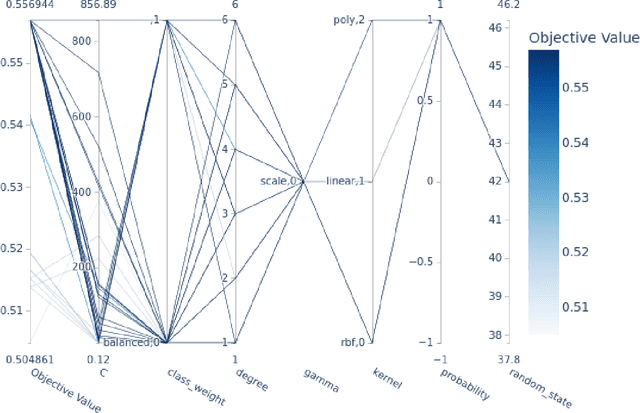
Machine learning (ML) offers powerful methods for detecting and modeling associations often in data with large feature spaces and complex associations. Many useful tools/packages (e.g. scikit-learn) have been developed to make the various elements of data handling, processing, modeling, and interpretation accessible. However, it is not trivial for most investigators to assemble these elements into a rigorous, replicatable, unbiased, and effective data analysis pipeline. Automated machine learning (AutoML) seeks to address these issues by simplifying the process of ML analysis for all. Here, we introduce STREAMLINE, a simple, transparent, end-to-end AutoML pipeline designed as a framework to easily conduct rigorous ML modeling and analysis (limited initially to binary classification). STREAMLINE is specifically designed to compare performance between datasets, ML algorithms, and other AutoML tools. It is unique among other autoML tools by offering a fully transparent and consistent baseline of comparison using a carefully designed series of pipeline elements including: (1) exploratory analysis, (2) basic data cleaning, (3) cross validation partitioning, (4) data scaling and imputation, (5) filter-based feature importance estimation, (6) collective feature selection, (7) ML modeling with `Optuna' hyperparameter optimization across 15 established algorithms (including less well-known Genetic Programming and rule-based ML), (8) evaluation across 16 classification metrics, (9) model feature importance estimation, (10) statistical significance comparisons, and (11) automatically exporting all results, plots, a PDF summary report, and models that can be easily applied to replication data.
A Rigorous Machine Learning Analysis Pipeline for Biomedical Binary Classification: Application in Pancreatic Cancer Nested Case-control Studies with Implications for Bias Assessments
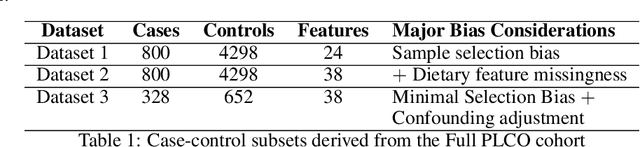
Sep 08, 2020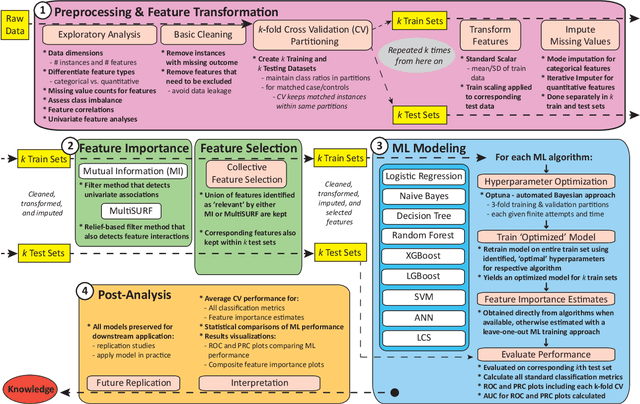
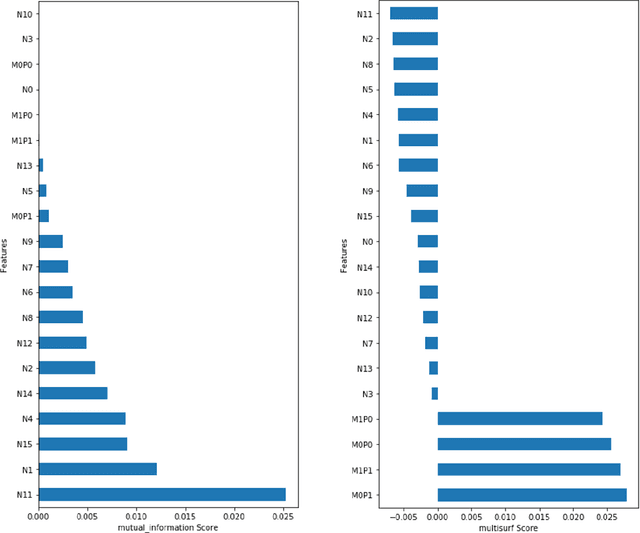
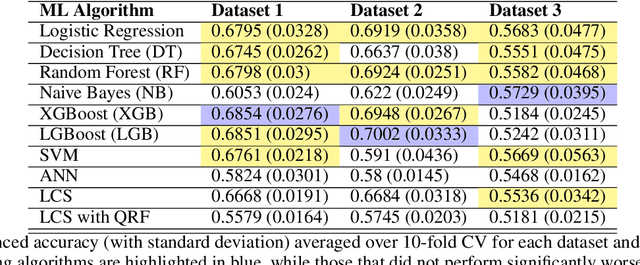
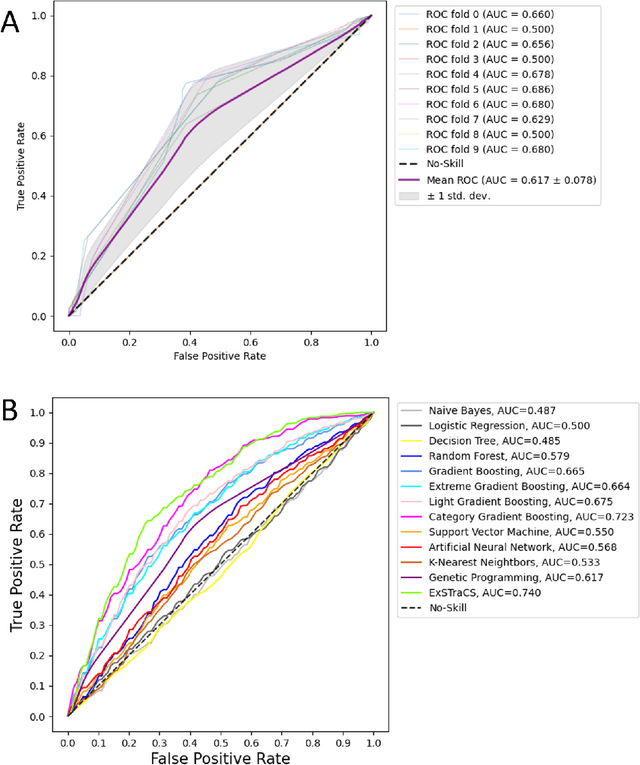
Machine learning (ML) offers a collection of powerful approaches for detecting and modeling associations, often applied to data having a large number of features and/or complex associations. Currently, there are many tools to facilitate implementing custom ML analyses (e.g. scikit-learn). Interest is also increasing in automated ML packages, which can make it easier for non-experts to apply ML and have the potential to improve model performance. ML permeates most subfields of biomedical research with varying levels of rigor and correct usage. Tremendous opportunities offered by ML are frequently offset by the challenge of assembling comprehensive analysis pipelines, and the ease of ML misuse. In this work we have laid out and assembled a complete, rigorous ML analysis pipeline focused on binary classification (i.e. case/control prediction), and applied this pipeline to both simulated and real world data. At a high level, this 'automated' but customizable pipeline includes a) exploratory analysis, b) data cleaning and transformation, c) feature selection, d) model training with 9 established ML algorithms, each with hyperparameter optimization, and e) thorough evaluation, including appropriate metrics, statistical analyses, and novel visualizations. This pipeline organizes the many subtle complexities of ML pipeline assembly to illustrate best practices to avoid bias and ensure reproducibility. Additionally, this pipeline is the first to compare established ML algorithms to 'ExSTraCS', a rule-based ML algorithm with the unique capability of interpretably modeling heterogeneous patterns of association. While designed to be widely applicable we apply this pipeline to an epidemiological investigation of established and newly identified risk factors for pancreatic cancer to evaluate how different sources of bias might be handled by ML algorithms.