 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLCS-DIVE: An Automated Rule-based Machine Learning Visualization Pipeline for Characterizing Complex Associations in Classification
Apr 26, 2021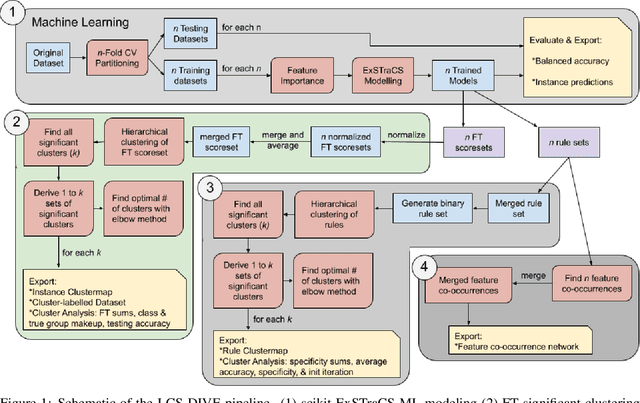
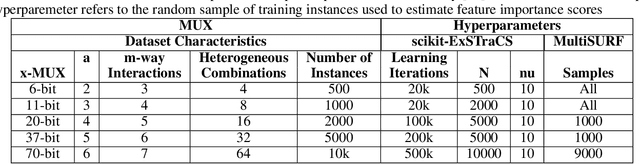
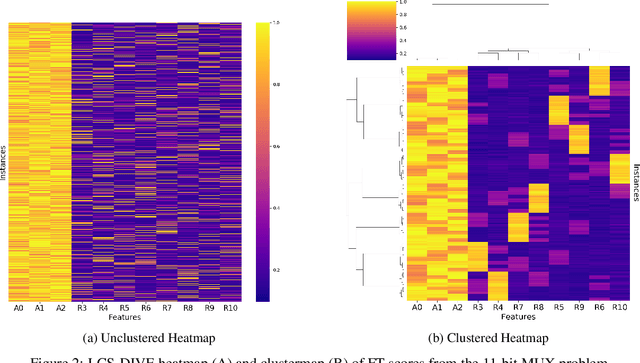
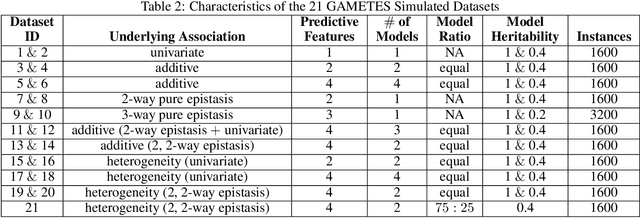
Machine learning (ML) research has yielded powerful tools for training accurate prediction models despite complex multivariate associations (e.g. interactions and heterogeneity). In fields such as medicine, improved interpretability of ML modeling is required for knowledge discovery, accountability, and fairness. Rule-based ML approaches such as Learning Classifier Systems (LCSs) strike a balance between predictive performance and interpretability in complex, noisy domains. This work introduces the LCS Discovery and Visualization Environment (LCS-DIVE), an automated LCS model interpretation pipeline for complex biomedical classification. LCS-DIVE conducts modeling using a new scikit-learn implementation of ExSTraCS, an LCS designed to overcome noise and scalability in biomedical data mining yielding human readable IF:THEN rules as well as feature-tracking scores for each training sample. LCS-DIVE leverages feature-tracking scores and/or rules to automatically guide characterization of (1) feature importance (2) underlying additive, epistatic, and/or heterogeneous patterns of association, and (3) model-driven heterogeneous instance subgroups via clustering, visualization generation, and cluster interrogation. LCS-DIVE was evaluated over a diverse set of simulated genetic and benchmark datasets encoding a variety of complex multivariate associations, demonstrating its ability to differentiate between them and then applied to characterize associations within a real-world study of pancreatic cancer.
A Rigorous Machine Learning Analysis Pipeline for Biomedical Binary Classification: Application in Pancreatic Cancer Nested Case-control Studies with Implications for Bias Assessments
Sep 08, 2020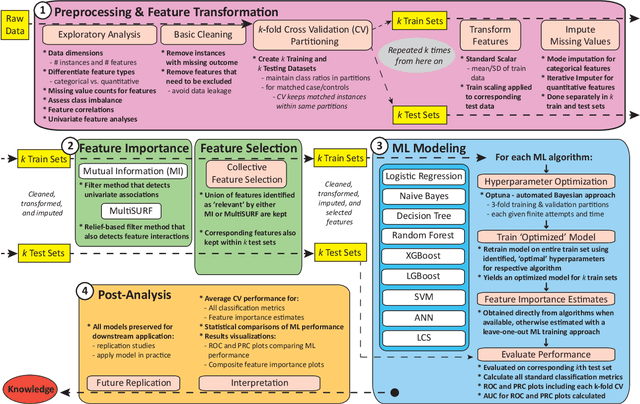
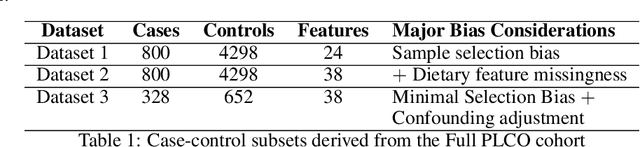
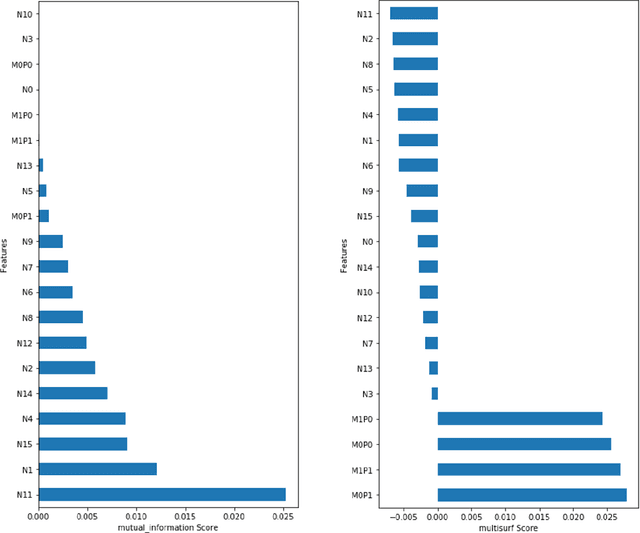
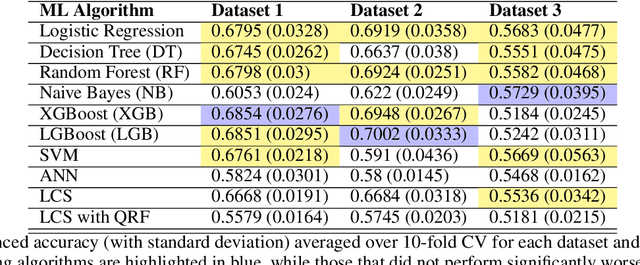
Machine learning (ML) offers a collection of powerful approaches for detecting and modeling associations, often applied to data having a large number of features and/or complex associations. Currently, there are many tools to facilitate implementing custom ML analyses (e.g. scikit-learn). Interest is also increasing in automated ML packages, which can make it easier for non-experts to apply ML and have the potential to improve model performance. ML permeates most subfields of biomedical research with varying levels of rigor and correct usage. Tremendous opportunities offered by ML are frequently offset by the challenge of assembling comprehensive analysis pipelines, and the ease of ML misuse. In this work we have laid out and assembled a complete, rigorous ML analysis pipeline focused on binary classification (i.e. case/control prediction), and applied this pipeline to both simulated and real world data. At a high level, this 'automated' but customizable pipeline includes a) exploratory analysis, b) data cleaning and transformation, c) feature selection, d) model training with 9 established ML algorithms, each with hyperparameter optimization, and e) thorough evaluation, including appropriate metrics, statistical analyses, and novel visualizations. This pipeline organizes the many subtle complexities of ML pipeline assembly to illustrate best practices to avoid bias and ensure reproducibility. Additionally, this pipeline is the first to compare established ML algorithms to 'ExSTraCS', a rule-based ML algorithm with the unique capability of interpretably modeling heterogeneous patterns of association. While designed to be widely applicable we apply this pipeline to an epidemiological investigation of established and newly identified risk factors for pancreatic cancer to evaluate how different sources of bias might be handled by ML algorithms.