 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAgentFactory: A Self-Evolving Framework Through Executable Subagent Accumulation and Reuse
Mar 18, 2026Building LLM-based agents has become increasingly important. Recent works on LLM-based agent self-evolution primarily record successful experiences as textual prompts or reflections, which cannot reliably guarantee efficient task re-execution in complex scenarios. We propose AgentFactory, a new self-evolution paradigm that preserves successful task solutions as executable subagent code rather than textual experience. Crucially, these subagents are continuously refined based on execution feedback, becoming increasingly robust and efficient as more tasks are encountered. Saved subagents are pure Python code with standardized documentation, enabling portability across any Python-capable system. We demonstrate that AgentFactory enables continuous capability accumulation: its library of executable subagents grows and improves over time, progressively reducing the effort required for similar tasks without manual intervention. Our implementation is open-sourced at https://github.com/zzatpku/AgentFactory, and our demonstration video is available at https://youtu.be/iKSsuAXJHW0.
Uni-3DAR: Unified 3D Generation and Understanding via Autoregression on Compressed Spatial Tokens
Mar 21, 2025Recent advancements in large language models and their multi-modal extensions have demonstrated the effectiveness of unifying generation and understanding through autoregressive next-token prediction. However, despite the critical role of 3D structural generation and understanding (3D GU) in AI for science, these tasks have largely evolved independently, with autoregressive methods remaining underexplored. To bridge this gap, we introduce Uni-3DAR, a unified framework that seamlessly integrates 3D GU tasks via autoregressive prediction. At its core, Uni-3DAR employs a novel hierarchical tokenization that compresses 3D space using an octree, leveraging the inherent sparsity of 3D structures. It then applies an additional tokenization for fine-grained structural details, capturing key attributes such as atom types and precise spatial coordinates in microscopic 3D structures. We further propose two optimizations to enhance efficiency and effectiveness. The first is a two-level subtree compression strategy, which reduces the octree token sequence by up to 8x. The second is a masked next-token prediction mechanism tailored for dynamically varying token positions, significantly boosting model performance. By combining these strategies, Uni-3DAR successfully unifies diverse 3D GU tasks within a single autoregressive framework. Extensive experiments across multiple microscopic 3D GU tasks, including molecules, proteins, polymers, and crystals, validate its effectiveness and versatility. Notably, Uni-3DAR surpasses previous state-of-the-art diffusion models by a substantial margin, achieving up to 256\% relative improvement while delivering inference speeds up to 21.8x faster. The code is publicly available at https://github.com/dptech-corp/Uni-3DAR.
Representation Learning, Large-Scale 3D Molecular Pretraining, Molecular Property
Mar 13, 2025Molecular pretrained representations (MPR) has emerged as a powerful approach for addressing the challenge of limited supervised data in applications such as drug discovery and material design. While early MPR methods relied on 1D sequences and 2D graphs, recent advancements have incorporated 3D conformational information to capture rich atomic interactions. However, these prior models treat molecules merely as discrete atom sets, overlooking the space surrounding them. We argue from a physical perspective that only modeling these discrete points is insufficient. We first present a simple yet insightful observation: naively adding randomly sampled virtual points beyond atoms can surprisingly enhance MPR performance. In light of this, we propose a principled framework that incorporates the entire 3D space spanned by molecules. We implement the framework via a novel Transformer-based architecture, dubbed SpaceFormer, with three key components: (1) grid-based space discretization; (2) grid sampling/merging; and (3) efficient 3D positional encoding. Extensive experiments show that SpaceFormer significantly outperforms previous 3D MPR models across various downstream tasks with limited data, validating the benefit of leveraging the additional 3D space beyond atoms in MPR models.
Multi-level protein pre-training with Vabs-Net
Feb 05, 2024In recent years, there has been a surge in the development of 3D structure-based pre-trained protein models, representing a significant advancement over pre-trained protein language models in various downstream tasks. However, most existing structure-based pre-trained models primarily focus on the residue level, i.e., alpha carbon atoms, while ignoring other atoms like side chain atoms. We argue that modeling proteins at both residue and atom levels is important since the side chain atoms can also be crucial for numerous downstream tasks, for example, molecular docking. Nevertheless, we find that naively combining residue and atom information during pre-training typically fails. We identify a key reason is the information leakage caused by the inclusion of atom structure in the input, which renders residue-level pre-training tasks trivial and results in insufficiently expressive residue representations. To address this issue, we introduce a span mask pre-training strategy on 3D protein chains to learn meaningful representations of both residues and atoms. This leads to a simple yet effective approach to learning protein representation suitable for diverse downstream tasks. Extensive experimental results on binding site prediction and function prediction tasks demonstrate our proposed pre-training approach significantly outperforms other methods. Our code will be made public.
End-to-End Crystal Structure Prediction from Powder X-Ray Diffraction
Jan 08, 2024



Powder X-ray diffraction (PXRD) is a crucial means for crystal structure determination. Such determination often involves external database matching to find a structural analogue and Rietveld refinement to obtain finer structure. However, databases may be incomplete and Rietveld refinement often requires intensive trial-and-error efforts from trained experimentalists, which remains ineffective in practice. To settle these issues, we propose XtalNet, the first end-to-end deep learning-based framework capable of ab initio generation of crystal structures that accurately match given PXRD patterns. The model employs contrastive learning and Diffusion-based conditional generation to enable the simultaneous execution of two tasks: crystal structure retrieval based on PXRD patterns and conditional structure generations. To validate the effectiveness of XtalNet, we curate a much more challenging and practical dataset hMOF-100, XtalNet performs well on this dataset, reaching 96.3\% top-10 hit ratio on the database retrieval task and 95.0\% top-10 match rate on the ranked structure generation task.
Highly Accurate Quantum Chemical Property Prediction with Uni-Mol+
Mar 16, 2023



Recent developments in deep learning have made remarkable progress in speeding up the prediction of quantum chemical (QC) properties by removing the need for expensive electronic structure calculations like density functional theory. However, previous methods that relied on 1D SMILES sequences or 2D molecular graphs failed to achieve high accuracy as QC properties are primarily dependent on the 3D equilibrium conformations optimized by electronic structure methods. In this paper, we propose a novel approach called Uni-Mol+ to tackle this challenge. Firstly, given a 2D molecular graph, Uni-Mol+ generates an initial 3D conformation from inexpensive methods such as RDKit. Then, the initial conformation is iteratively optimized to its equilibrium conformation, and the optimized conformation is further used to predict the QC properties. All these steps are automatically learned using Transformer models. We observed the quality of the optimized conformation is crucial for QC property prediction performance. To effectively optimize conformation, we introduce a two-track Transformer model backbone in Uni-Mol+ and train it together with the QC property prediction task. We also design a novel training approach called linear trajectory injection to ensure proper supervision for the Uni-Mol+ learning process. Our extensive benchmarking results demonstrate that the proposed Uni-Mol+ significantly improves the accuracy of QC property prediction. We have made the code and model publicly available at \url{https://github.com/dptech-corp/Uni-Mol}.
Do Deep Learning Models Really Outperform Traditional Approaches in Molecular Docking?
Feb 23, 2023
Molecular docking, given a ligand molecule and a ligand binding site (called ``pocket'') on a protein, predicting the binding mode of the protein-ligand complex, is a widely used technique in drug design. Many deep learning models have been developed for molecular docking, while most existing deep learning models perform docking on the whole protein, rather than on a given pocket as the traditional molecular docking approaches, which does not match common needs. What's more, they claim to perform better than traditional molecular docking, but the approach of comparison is not fair, since traditional methods are not designed for docking on the whole protein without a given pocket. In this paper, we design a series of experiments to examine the actual performance of these deep learning models and traditional methods. For a fair comparison, we decompose the docking on the whole protein into two steps, pocket searching and docking on a given pocket, and build pipelines to evaluate traditional methods and deep learning methods respectively. We find that deep learning models are actually good at pocket searching, but traditional methods are better than deep learning models at docking on given pockets. Overall, our work explicitly reveals some potential problems in current deep learning models for molecular docking and provides several suggestions for future works.
3D Molecular Generation via Virtual Dynamics
Feb 12, 2023



Structure-based drug design, i.e., finding molecules with high affinities to the target protein pocket, is one of the most critical tasks in drug discovery. Traditional solutions, like virtual screening, require exhaustively searching on a large molecular database, which are inefficient and cannot return novel molecules beyond the database. The pocket-based 3D molecular generation model, i.e., directly generating a molecule with a 3D structure and binding position in the pocket, is a new promising way to address this issue. Herein, we propose VD-Gen, a novel pocket-based 3D molecular generation pipeline. VD-Gen consists of several carefully designed stages to generate fine-grained 3D molecules with binding positions in the pocket cavity end-to-end. Rather than directly generating or sampling atoms with 3D positions in the pocket like in early attempts, in VD-Gen, we first randomly initialize many virtual particles in the pocket; then iteratively move these virtual particles, making the distribution of virtual particles approximate the distribution of molecular atoms. After virtual particles are stabilized in 3D space, we extract a 3D molecule from them. Finally, we further refine atoms in the extracted molecule by iterative movement again, to get a high-quality 3D molecule, and predict a confidence score for it. Extensive experiment results on pocket-based molecular generation demonstrate that VD-Gen can generate novel 3D molecules to fill the target pocket cavity with high binding affinities, significantly outperforming previous baselines.
Less is More: Pre-training a Strong Siamese Encoder Using a Weak Decoder
Feb 18, 2021
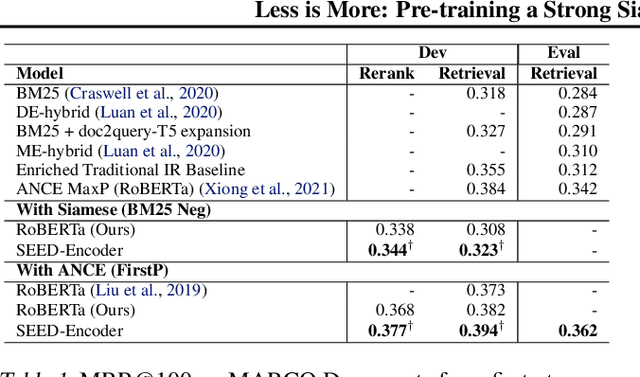
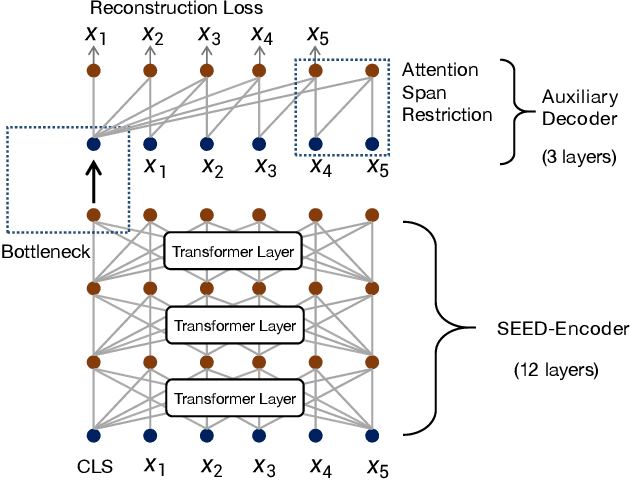
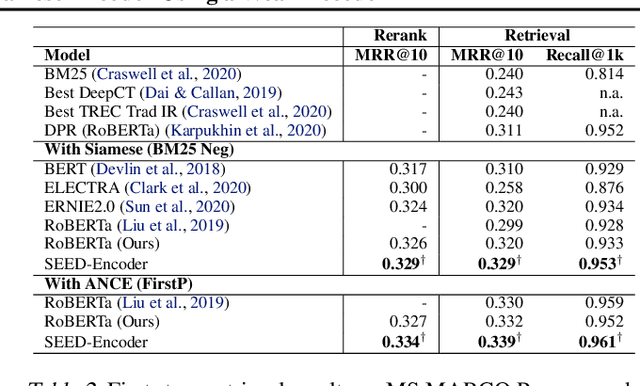
Many real-world applications use Siamese networks to efficiently match text sequences at scale, which require high-quality sequence encodings. This paper pre-trains language models dedicated to sequence matching in Siamese architectures. We first hypothesize that a representation is better for sequence matching if the entire sequence can be reconstructed from it, which, however, is unlikely to be achieved in standard autoencoders: A strong decoder can rely on its capacity and natural language patterns to reconstruct and bypass the needs of better sequence encodings. Therefore we propose a new self-learning method that pretrains the encoder with a weak decoder, which reconstructs the original sequence from the encoder's [CLS] representations but is restricted in both capacity and attention span. In our experiments on web search and recommendation, the pre-trained SEED-Encoder, "SiamEsE oriented encoder by reconstructing from weak decoder", shows significantly better generalization ability when fine-tuned in Siamese networks, improving overall accuracy and few-shot performances. Our code and models will be released.