 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBenchmarking Relief-Based Feature Selection Methods for Bioinformatics Data Mining
Apr 03, 2018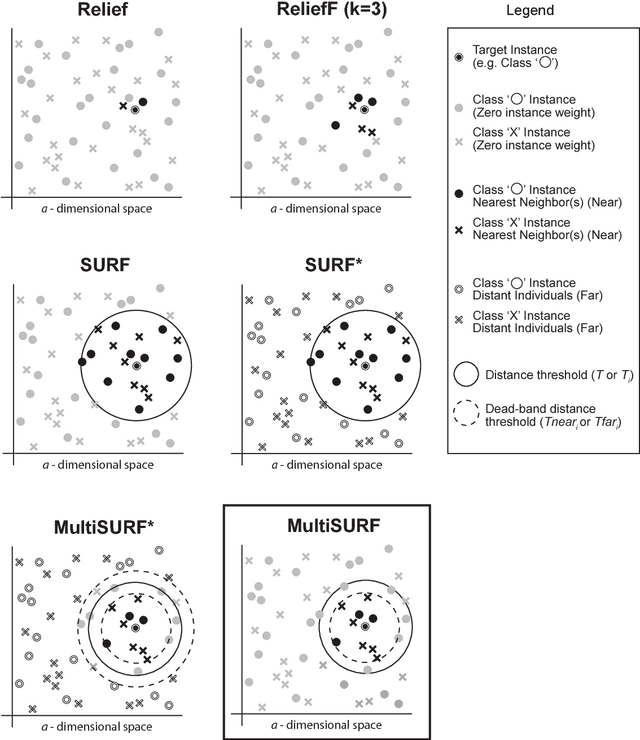
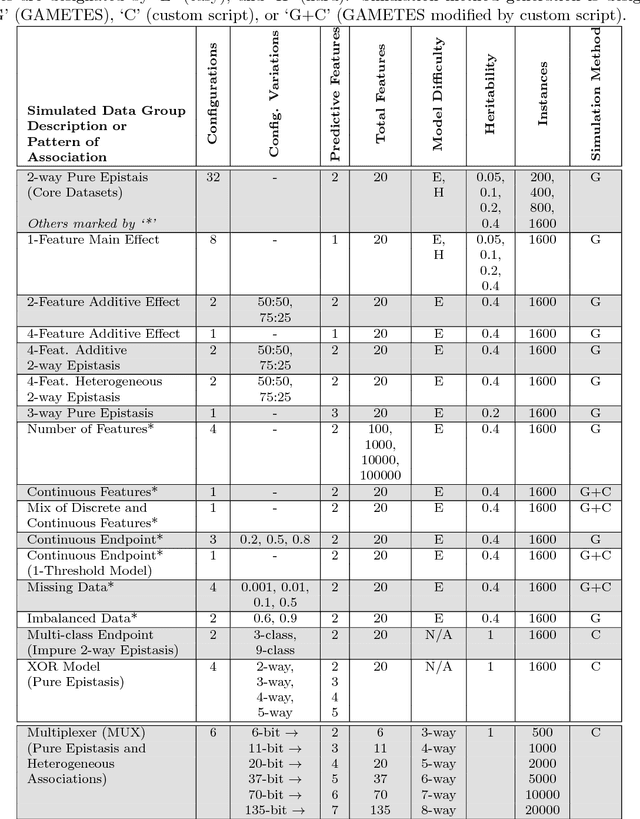
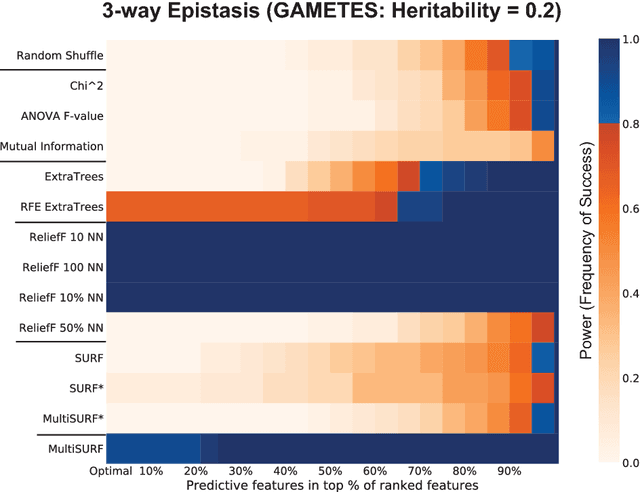
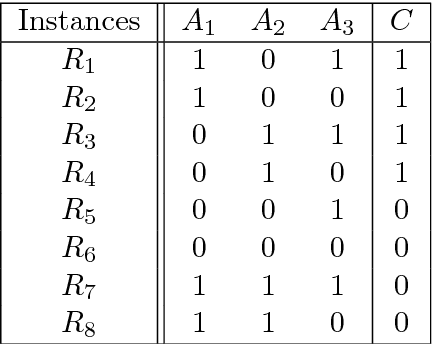
Modern biomedical data mining requires feature selection methods that can (1) be applied to large scale feature spaces (e.g. `omics' data), (2) function in noisy problems, (3) detect complex patterns of association (e.g. gene-gene interactions), (4) be flexibly adapted to various problem domains and data types (e.g. genetic variants, gene expression, and clinical data) and (5) are computationally tractable. To that end, this work examines a set of filter-style feature selection algorithms inspired by the `Relief' algorithm, i.e. Relief-Based algorithms (RBAs). We implement and expand these RBAs in an open source framework called ReBATE (Relief-Based Algorithm Training Environment). We apply a comprehensive genetic simulation study comparing existing RBAs, a proposed RBA called MultiSURF, and other established feature selection methods, over a variety of problems. The results of this study (1) support the assertion that RBAs are particularly flexible, efficient, and powerful feature selection methods that differentiate relevant features having univariate, multivariate, epistatic, or heterogeneous associations, (2) confirm the efficacy of expansions for classification vs. regression, discrete vs. continuous features, missing data, multiple classes, or class imbalance, (3) identify previously unknown limitations of specific RBAs, and (4) suggest that while MultiSURF* performs best for explicitly identifying pure 2-way interactions, MultiSURF yields the most reliable feature selection performance across a wide range of problem types.
Relief-Based Feature Selection: Introduction and Review
Apr 02, 2018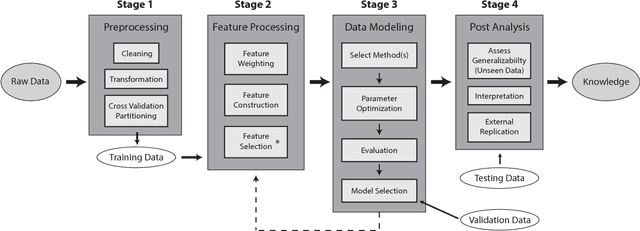
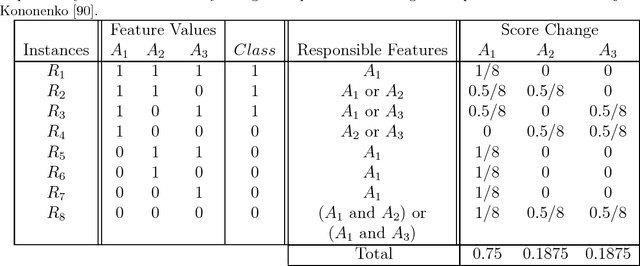
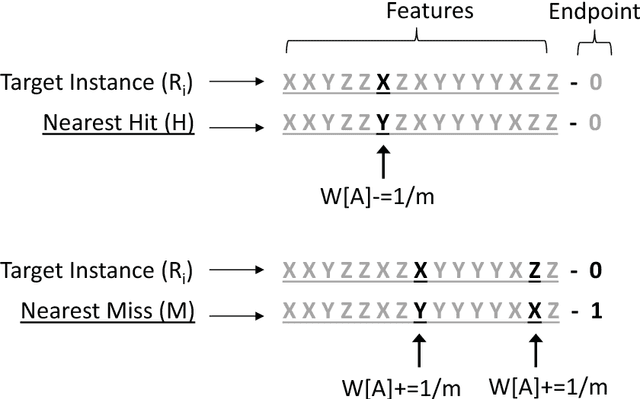

Feature selection plays a critical role in biomedical data mining, driven by increasing feature dimensionality in target problems and growing interest in advanced but computationally expensive methodologies able to model complex associations. Specifically, there is a need for feature selection methods that are computationally efficient, yet sensitive to complex patterns of association, e.g. interactions, so that informative features are not mistakenly eliminated prior to downstream modeling. This paper focuses on Relief-based algorithms (RBAs), a unique family of filter-style feature selection algorithms that have gained appeal by striking an effective balance between these objectives while flexibly adapting to various data characteristics, e.g. classification vs. regression. First, this work broadly examines types of feature selection and defines RBAs within that context. Next, we introduce the original Relief algorithm and associated concepts, emphasizing the intuition behind how it works, how feature weights generated by the algorithm can be interpreted, and why it is sensitive to feature interactions without evaluating combinations of features. Lastly, we include an expansive review of RBA methodological research beyond Relief and its popular descendant, ReliefF. In particular, we characterize branches of RBA research, and provide comparative summaries of RBA algorithms including contributions, strategies, functionality, time complexity, adaptation to key data characteristics, and software availability.
Layered TPOT: Speeding up Tree-based Pipeline Optimization
Mar 12, 2018
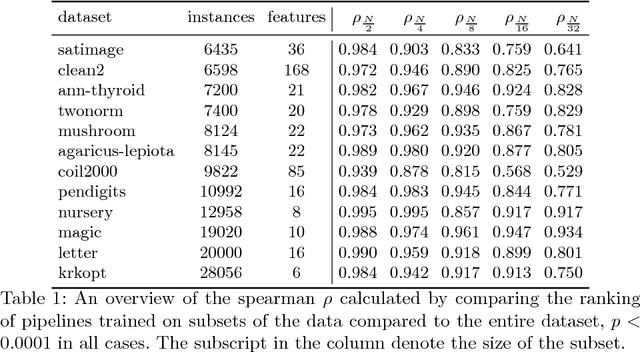
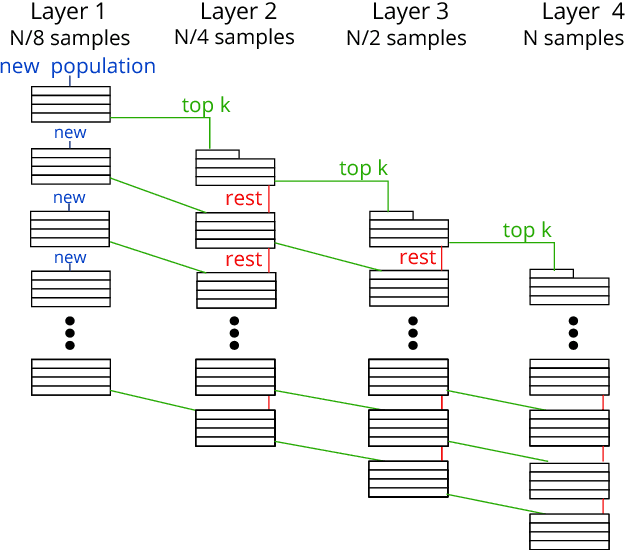
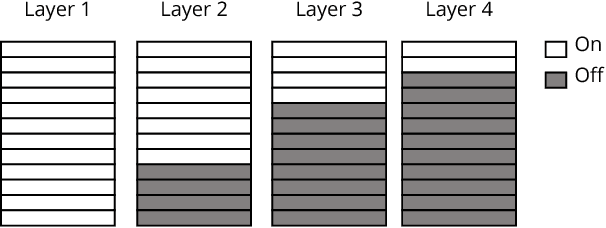
With the demand for machine learning increasing, so does the demand for tools which make it easier to use. Automated machine learning (AutoML) tools have been developed to address this need, such as the Tree-Based Pipeline Optimization Tool (TPOT) which uses genetic programming to build optimal pipelines. We introduce Layered TPOT, a modification to TPOT which aims to create pipelines equally good as the original, but in significantly less time. This approach evaluates candidate pipelines on increasingly large subsets of the data according to their fitness, using a modified evolutionary algorithm to allow for separate competition between pipelines trained on different sample sizes. Empirical evaluation shows that, on sufficiently large datasets, Layered TPOT indeed finds better models faster.
Data-driven Advice for Applying Machine Learning to Bioinformatics Problems
Jan 07, 2018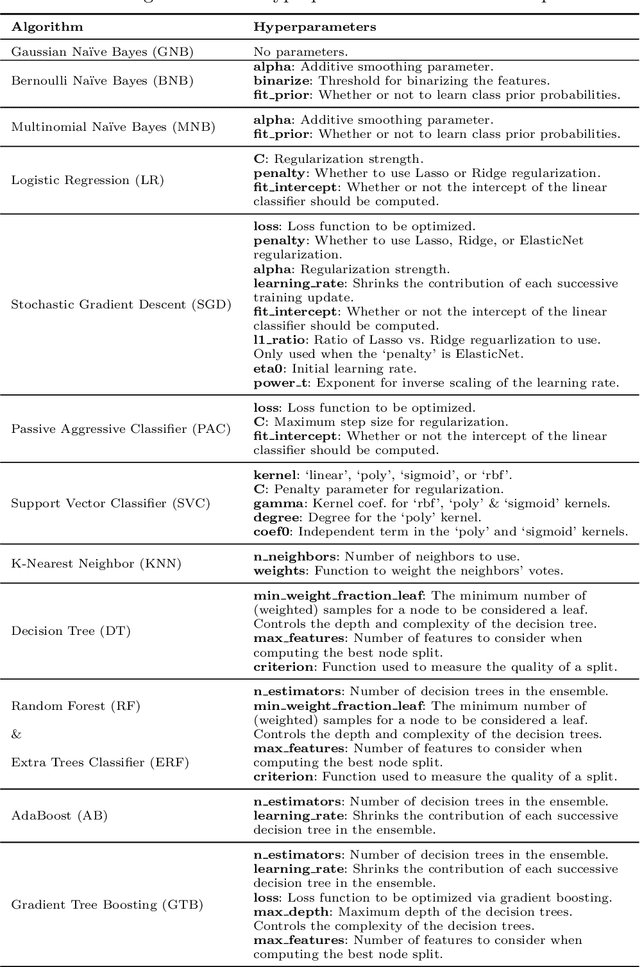
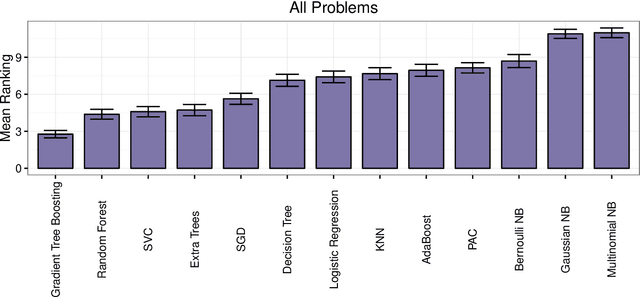
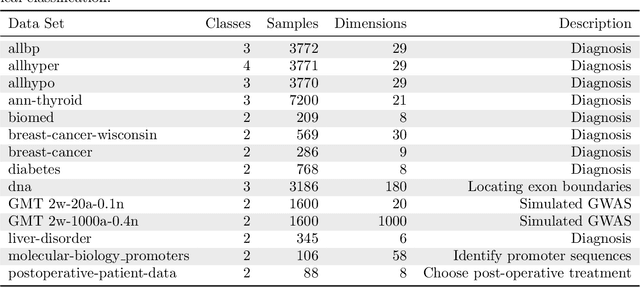
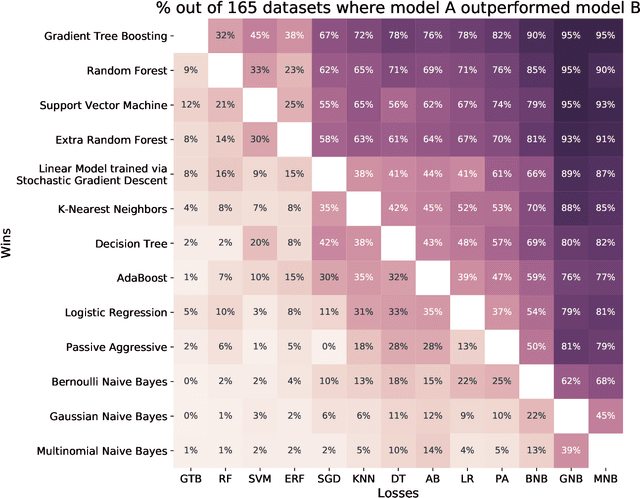
As the bioinformatics field grows, it must keep pace not only with new data but with new algorithms. Here we contribute a thorough analysis of 13 state-of-the-art, commonly used machine learning algorithms on a set of 165 publicly available classification problems in order to provide data-driven algorithm recommendations to current researchers. We present a number of statistical and visual comparisons of algorithm performance and quantify the effect of model selection and algorithm tuning for each algorithm and dataset. The analysis culminates in the recommendation of five algorithms with hyperparameters that maximize classifier performance across the tested problems, as well as general guidelines for applying machine learning to supervised classification problems.
Considerations of automated machine learning in clinical metabolic profiling: Altered homocysteine plasma concentration associated with metformin exposure
Oct 09, 2017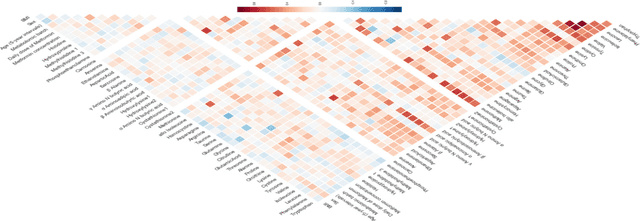
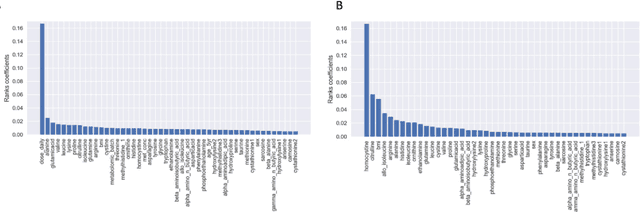
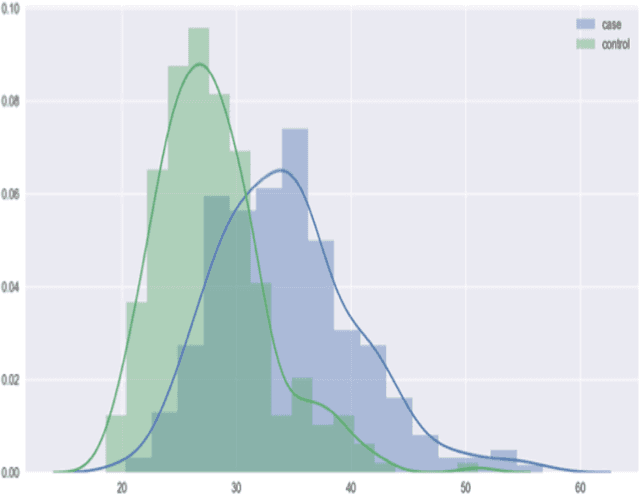
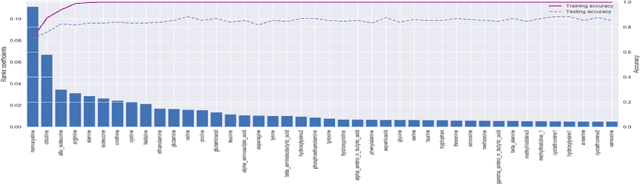
With the maturation of metabolomics science and proliferation of biobanks, clinical metabolic profiling is an increasingly opportunistic frontier for advancing translational clinical research. Automated Machine Learning (AutoML) approaches provide exciting opportunity to guide feature selection in agnostic metabolic profiling endeavors, where potentially thousands of independent data points must be evaluated. In previous research, AutoML using high-dimensional data of varying types has been demonstrably robust, outperforming traditional approaches. However, considerations for application in clinical metabolic profiling remain to be evaluated. Particularly, regarding the robustness of AutoML to identify and adjust for common clinical confounders. In this study, we present a focused case study regarding AutoML considerations for using the Tree-Based Optimization Tool (TPOT) in metabolic profiling of exposure to metformin in a biobank cohort. First, we propose a tandem rank-accuracy measure to guide agnostic feature selection and corresponding threshold determination in clinical metabolic profiling endeavors. Second, while AutoML, using default parameters, demonstrated potential to lack sensitivity to low-effect confounding clinical covariates, we demonstrated residual training and adjustment of metabolite features as an easily applicable approach to ensure AutoML adjustment for potential confounding characteristics. Finally, we present increased homocysteine with long-term exposure to metformin as a potentially novel, non-replicated metabolite association suggested by TPOT; an association not identified in parallel clinical metabolic profiling endeavors. While considerations are recommended, including adjustment approaches for clinical confounders, AutoML presents an exciting tool to enhance clinical metabolic profiling and advance translational research endeavors.
* Manuscript - containing supplementary information - accepted (9/15/2017) for publication within Pacific Symposium on Biocomputing 2018 <https://psb.stanford.edu/psb-online>. Original supplementary information includes an additional 6 pages of content (18 pages total) and 8 figures (13 figures total)
Markov Brains: A Technical Introduction
Sep 17, 2017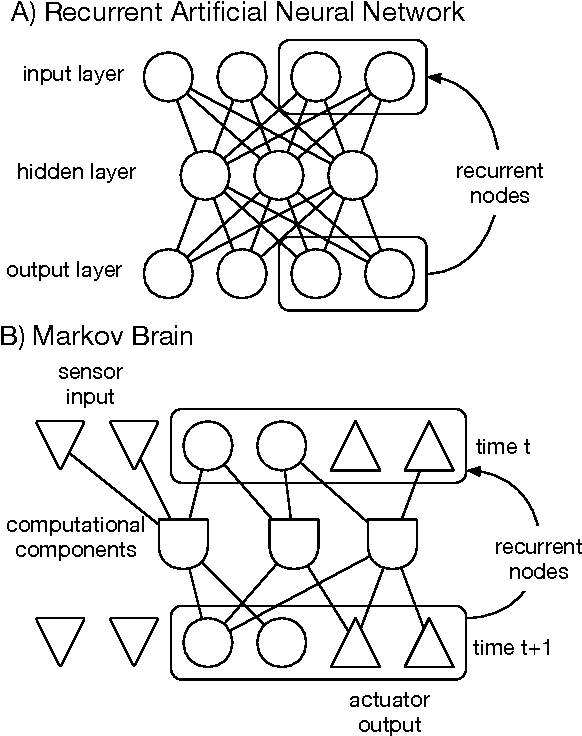
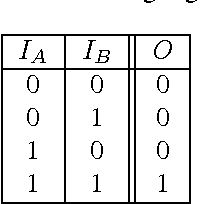
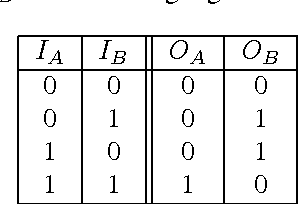

Markov Brains are a class of evolvable artificial neural networks (ANN). They differ from conventional ANNs in many aspects, but the key difference is that instead of a layered architecture, with each node performing the same function, Markov Brains are networks built from individual computational components. These computational components interact with each other, receive inputs from sensors, and control motor outputs. The function of the computational components, their connections to each other, as well as connections to sensors and motors are all subject to evolutionary optimization. Here we describe in detail how a Markov Brain works, what techniques can be used to study them, and how they can be evolved.
A System for Accessible Artificial Intelligence
Aug 10, 2017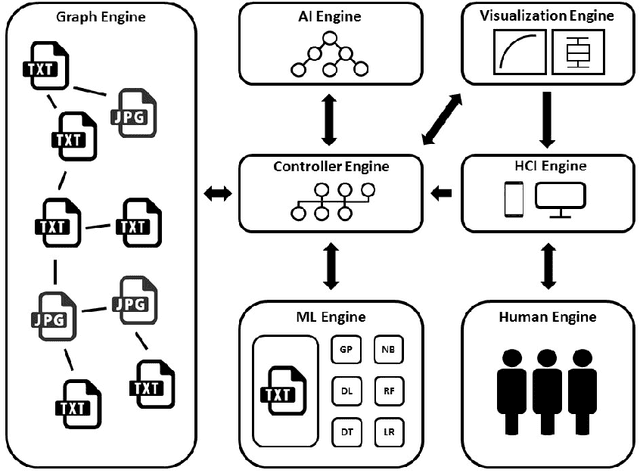


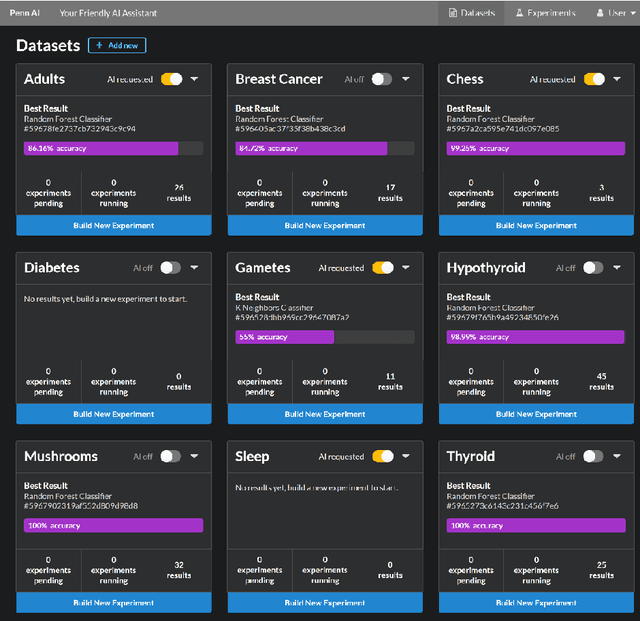
While artificial intelligence (AI) has become widespread, many commercial AI systems are not yet accessible to individual researchers nor the general public due to the deep knowledge of the systems required to use them. We believe that AI has matured to the point where it should be an accessible technology for everyone. We present an ongoing project whose ultimate goal is to deliver an open source, user-friendly AI system that is specialized for machine learning analysis of complex data in the biomedical and health care domains. We discuss how genetic programming can aid in this endeavor, and highlight specific examples where genetic programming has automated machine learning analyses in previous projects.
PMLB: A Large Benchmark Suite for Machine Learning Evaluation and Comparison
Mar 01, 2017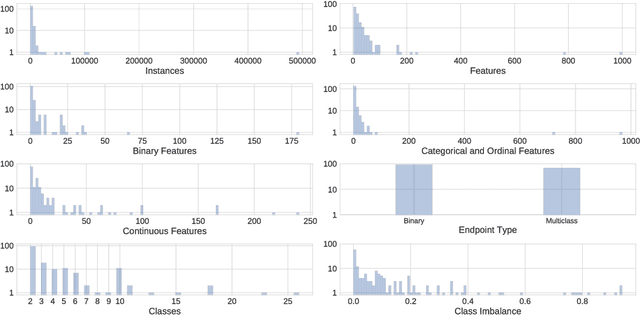
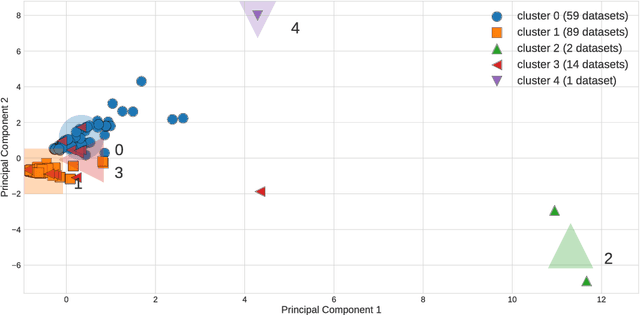
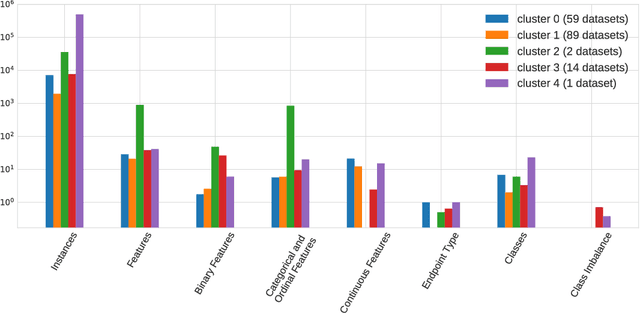
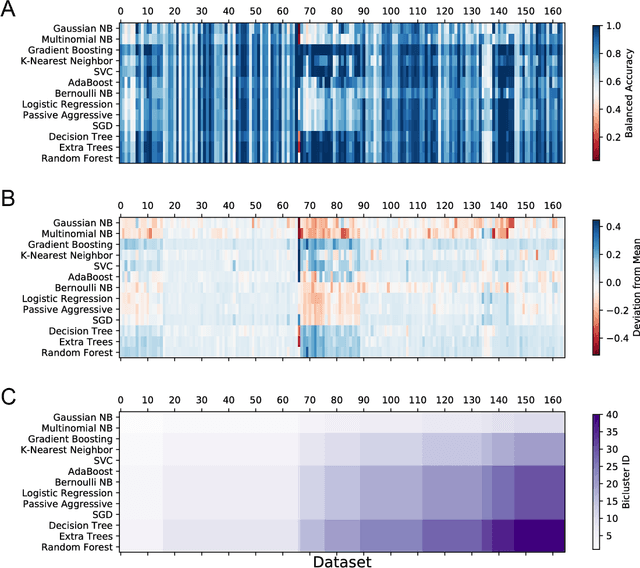
The selection, development, or comparison of machine learning methods in data mining can be a difficult task based on the target problem and goals of a particular study. Numerous publicly available real-world and simulated benchmark datasets have emerged from different sources, but their organization and adoption as standards have been inconsistent. As such, selecting and curating specific benchmarks remains an unnecessary burden on machine learning practitioners and data scientists. The present study introduces an accessible, curated, and developing public benchmark resource to facilitate identification of the strengths and weaknesses of different machine learning methodologies. We compare meta-features among the current set of benchmark datasets in this resource to characterize the diversity of available data. Finally, we apply a number of established machine learning methods to the entire benchmark suite and analyze how datasets and algorithms cluster in terms of performance. This work is an important first step towards understanding the limitations of popular benchmarking suites and developing a resource that connects existing benchmarking standards to more diverse and efficient standards in the future.
Toward the automated analysis of complex diseases in genome-wide association studies using genetic programming
Feb 06, 2017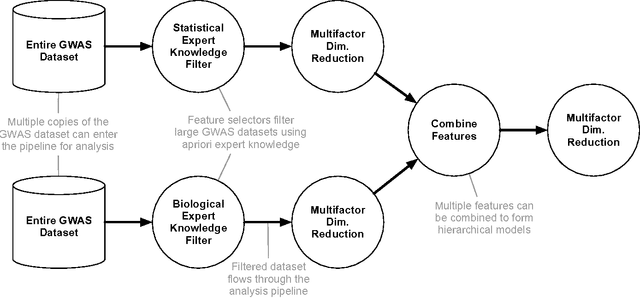
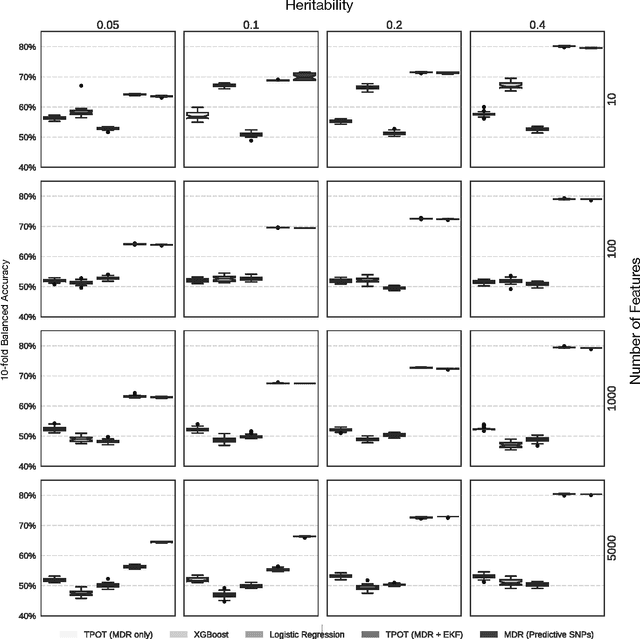
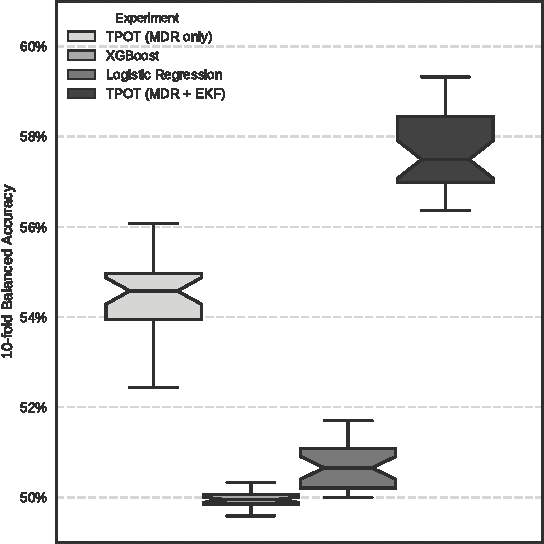
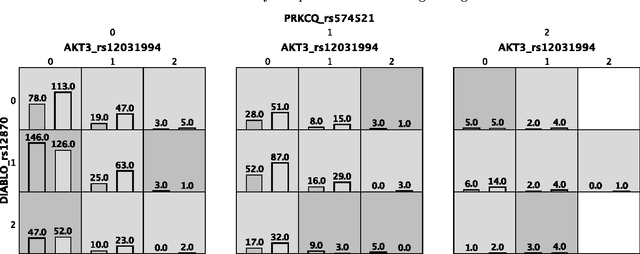
Machine learning has been gaining traction in recent years to meet the demand for tools that can efficiently analyze and make sense of the ever-growing databases of biomedical data in health care systems around the world. However, effectively using machine learning methods requires considerable domain expertise, which can be a barrier of entry for bioinformaticians new to computational data science methods. Therefore, off-the-shelf tools that make machine learning more accessible can prove invaluable for bioinformaticians. To this end, we have developed an open source pipeline optimization tool (TPOT-MDR) that uses genetic programming to automatically design machine learning pipelines for bioinformatics studies. In TPOT-MDR, we implement Multifactor Dimensionality Reduction (MDR) as a feature construction method for modeling higher-order feature interactions, and combine it with a new expert knowledge-guided feature selector for large biomedical data sets. We demonstrate TPOT-MDR's capabilities using a combination of simulated and real world data sets from human genetics and find that TPOT-MDR significantly outperforms modern machine learning methods such as logistic regression and eXtreme Gradient Boosting (XGBoost). We further analyze the best pipeline discovered by TPOT-MDR for a real world problem and highlight TPOT-MDR's ability to produce a high-accuracy solution that is also easily interpretable.
Identifying and Harnessing the Building Blocks of Machine Learning Pipelines for Sensible Initialization of a Data Science Automation Tool
Jul 29, 2016

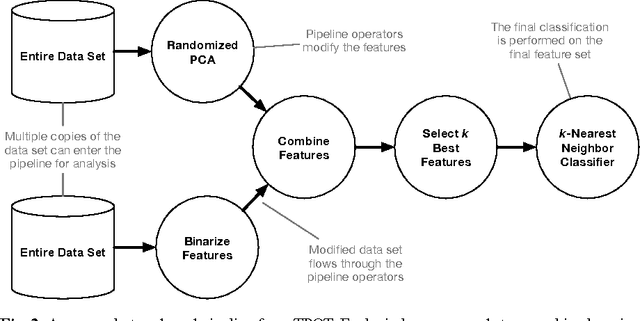

As data science continues to grow in popularity, there will be an increasing need to make data science tools more scalable, flexible, and accessible. In particular, automated machine learning (AutoML) systems seek to automate the process of designing and optimizing machine learning pipelines. In this chapter, we present a genetic programming-based AutoML system called TPOT that optimizes a series of feature preprocessors and machine learning models with the goal of maximizing classification accuracy on a supervised classification problem. Further, we analyze a large database of pipelines that were previously used to solve various supervised classification problems and identify 100 short series of machine learning operations that appear the most frequently, which we call the building blocks of machine learning pipelines. We harness these building blocks to initialize TPOT with promising solutions, and find that this sensible initialization method significantly improves TPOT's performance on one benchmark at no cost of significantly degrading performance on the others. Thus, sensible initialization with machine learning pipeline building blocks shows promise for GP-based AutoML systems, and should be further refined in future work.