 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeClustering Alzheimer's Disease Subtypes via Similarity Learning and Graph Diffusion
Oct 04, 2024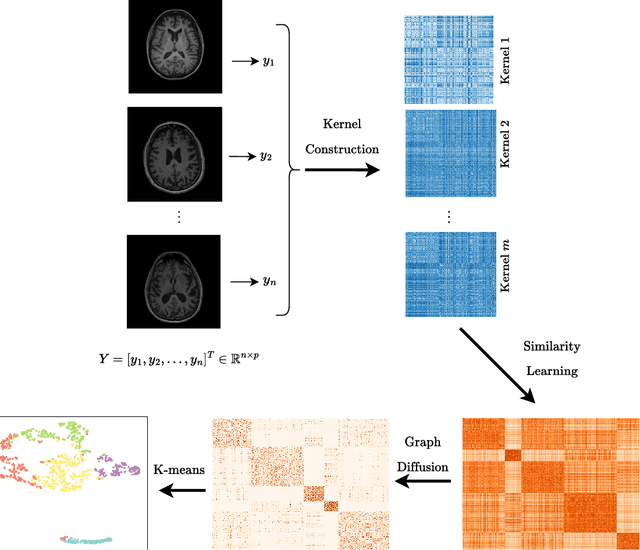
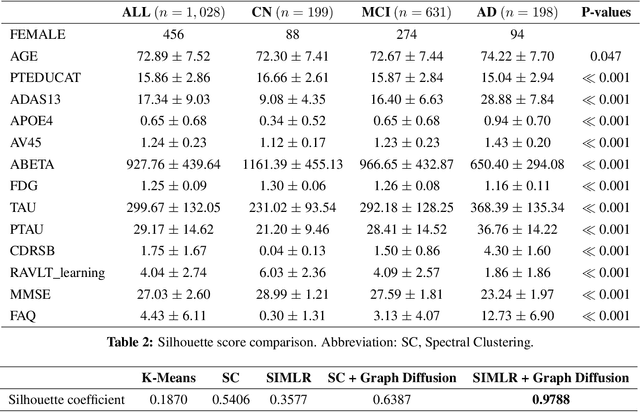
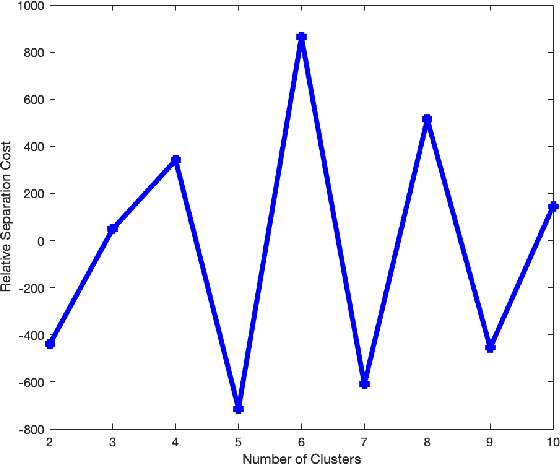
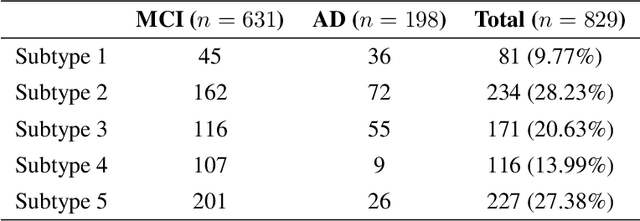
Alzheimer's disease (AD) is a complex neurodegenerative disorder that affects millions of people worldwide. Due to the heterogeneous nature of AD, its diagnosis and treatment pose critical challenges. Consequently, there is a growing research interest in identifying homogeneous AD subtypes that can assist in addressing these challenges in recent years. In this study, we aim to identify subtypes of AD that represent distinctive clinical features and underlying pathology by utilizing unsupervised clustering with graph diffusion and similarity learning. We adopted SIMLR, a multi-kernel similarity learning framework, and graph diffusion to perform clustering on a group of 829 patients with AD and mild cognitive impairment (MCI, a prodromal stage of AD) based on their cortical thickness measurements extracted from magnetic resonance imaging (MRI) scans. Although the clustering approach we utilized has not been explored for the task of AD subtyping before, it demonstrated significantly better performance than several commonly used clustering methods. Specifically, we showed the power of graph diffusion in reducing the effects of noise in the subtype detection. Our results revealed five subtypes that differed remarkably in their biomarkers, cognitive status, and some other clinical features. To evaluate the resultant subtypes further, a genetic association study was carried out and successfully identified potential genetic underpinnings of different AD subtypes. Our source code is available at: https://github.com/PennShenLab/AD-SIMLR.
Benchmarking AutoML algorithms on a collection of synthetic classification problems
Dec 14, 2022Automated machine learning (AutoML) algorithms have grown in popularity due to their high performance and flexibility to adapt to different problems and data sets. With the increasing number of AutoML algorithms, deciding which would best suit a given problem becomes increasingly more work. Therefore, it is essential to use complex and challenging benchmarks which would be able to differentiate the AutoML algorithms from each other. This paper compares the performance of four different AutoML algorithms: Tree-based Pipeline Optimization Tool (TPOT), Auto-Sklearn, Auto-Sklearn 2, and H2O AutoML. We use the Diverse and Generative ML benchmark (DIGEN), a diverse set of synthetic datasets derived from generative functions designed to highlight the strengths and weaknesses of the performance of common machine learning algorithms. We confirm that AutoML can identify pipelines that perform well on all included datasets. Most AutoML algorithms performed similarly without much room for improvement; however, some were more consistent than others at finding high-performing solutions for some datasets.
Applying Autonomous Hybrid Agent-based Computing to Difficult Optimization Problems
Oct 24, 2022
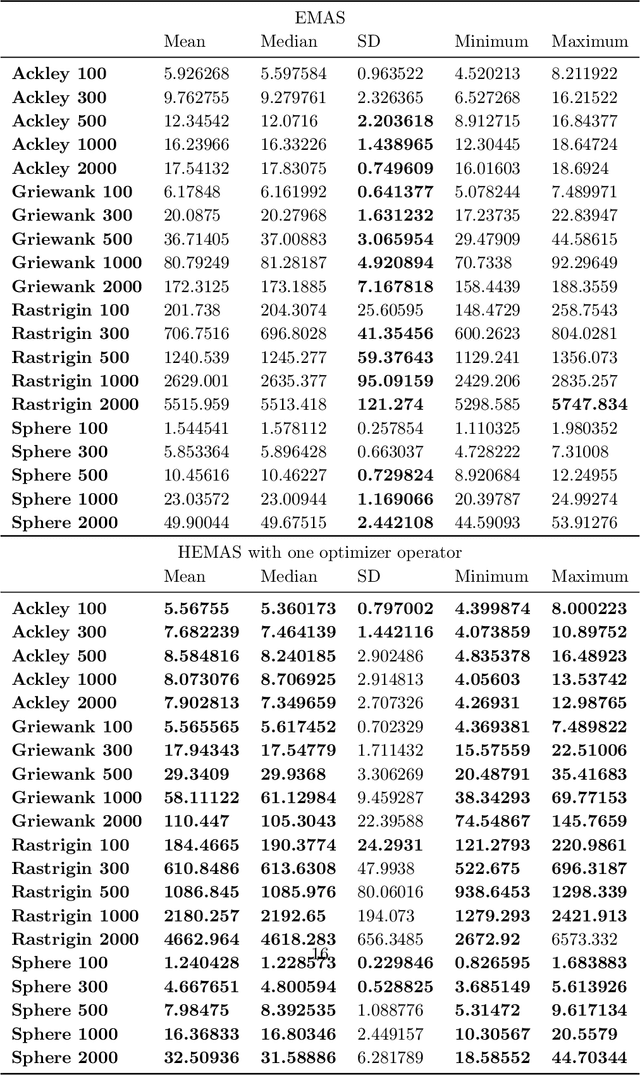
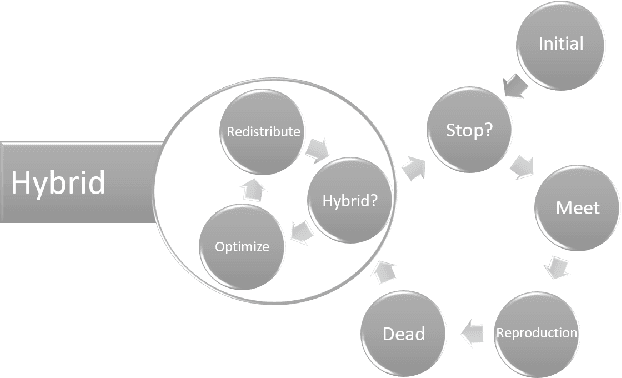
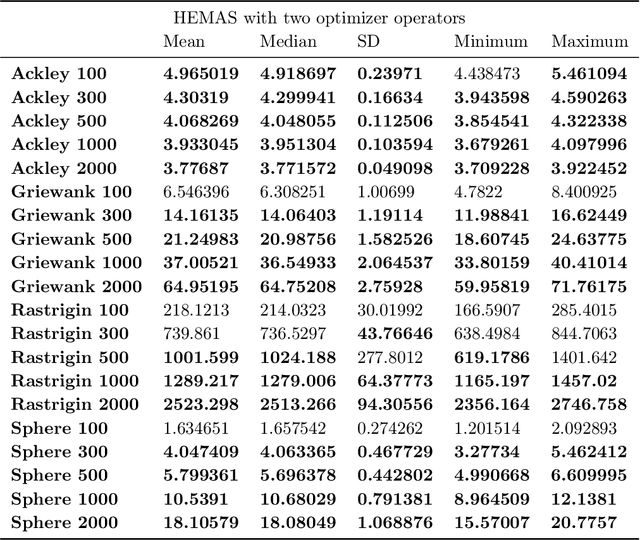
Evolutionary multi-agent systems (EMASs) are very good at dealing with difficult, multi-dimensional problems, their efficacy was proven theoretically based on analysis of the relevant Markov-Chain based model. Now the research continues on introducing autonomous hybridization into EMAS. This paper focuses on a proposed hybrid version of the EMAS, and covers selection and introduction of a number of hybrid operators and defining rules for starting the hybrid steps of the main algorithm. Those hybrid steps leverage existing, well-known and proven to be efficient metaheuristics, and integrate their results into the main algorithm. The discussed modifications are evaluated based on a number of difficult continuous-optimization benchmarks.
Contemporary Symbolic Regression Methods and their Relative Performance
Jul 29, 2021



Many promising approaches to symbolic regression have been presented in recent years, yet progress in the field continues to suffer from a lack of uniform, robust, and transparent benchmarking standards. In this paper, we address this shortcoming by introducing an open-source, reproducible benchmarking platform for symbolic regression. We assess 14 symbolic regression methods and 7 machine learning methods on a set of 252 diverse regression problems. Our assessment includes both real-world datasets with no known model form as well as ground-truth benchmark problems, including physics equations and systems of ordinary differential equations. For the real-world datasets, we benchmark the ability of each method to learn models with low error and low complexity relative to state-of-the-art machine learning methods. For the synthetic problems, we assess each method's ability to find exact solutions in the presence of varying levels of noise. Under these controlled experiments, we conclude that the best performing methods for real-world regression combine genetic algorithms with parameter estimation and/or semantic search drivers. When tasked with recovering exact equations in the presence of noise, we find that deep learning and genetic algorithm-based approaches perform similarly. We provide a detailed guide to reproducing this experiment and contributing new methods, and encourage other researchers to collaborate with us on a common and living symbolic regression benchmark.
Generative and reproducible benchmarks for comprehensive evaluation of machine learning classifiers
Jul 14, 2021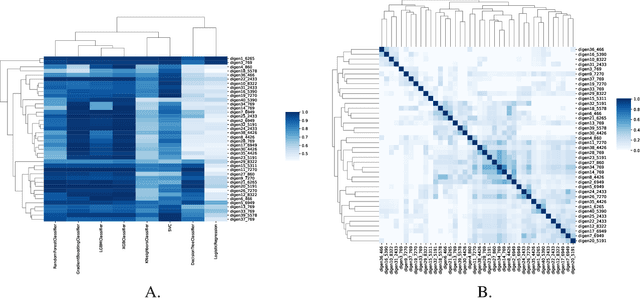

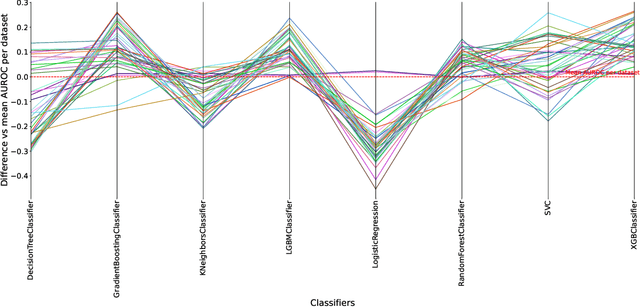
Understanding the strengths and weaknesses of machine learning (ML) algorithms is crucial for determine their scope of application. Here, we introduce the DIverse and GENerative ML Benchmark (DIGEN) - a collection of synthetic datasets for comprehensive, reproducible, and interpretable benchmarking of machine learning algorithms for classification of binary outcomes. The DIGEN resource consists of 40 mathematical functions which map continuous features to discrete endpoints for creating synthetic datasets. These 40 functions were discovered using a heuristic algorithm designed to maximize the diversity of performance among multiple popular machine learning algorithms thus providing a useful test suite for evaluating and comparing new methods. Access to the generative functions facilitates understanding of why a method performs poorly compared to other algorithms thus providing ideas for improvement. The resource with extensive documentation and analyses is open-source and available on GitHub.
EBIC.JL -- an Efficient Implementation of Evolutionary Biclustering Algorithm in Julia
May 03, 2021
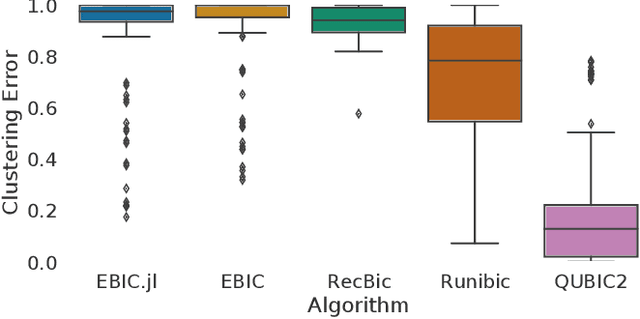
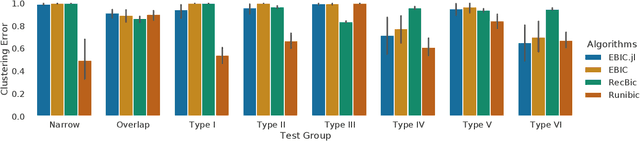
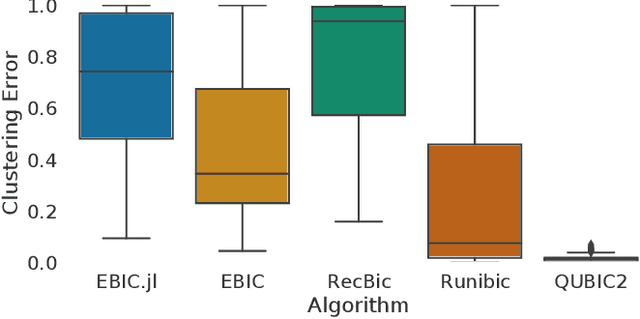
Biclustering is a data mining technique which searches for local patterns in numeric tabular data with main application in bioinformatics. This technique has shown promise in multiple areas, including development of biomarkers for cancer, disease subtype identification, or gene-drug interactions among others. In this paper we introduce EBIC.JL - an implementation of one of the most accurate biclustering algorithms in Julia, a modern highly parallelizable programming language for data science. We show that the new version maintains comparable accuracy to its predecessor EBIC while converging faster for the majority of the problems. We hope that this open source software in a high-level programming language will foster research in this promising field of bioinformatics and expedite development of new biclustering methods for big data.
Benchmarking in Optimization: Best Practice and Open Issues
Jul 07, 2020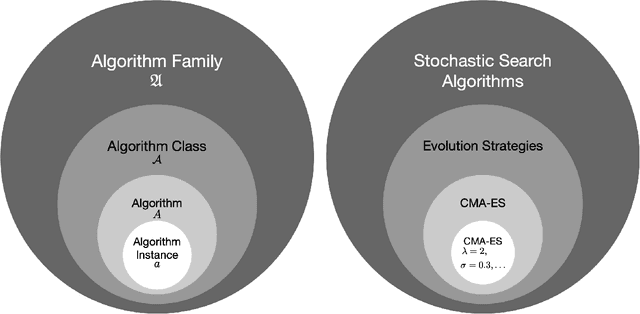
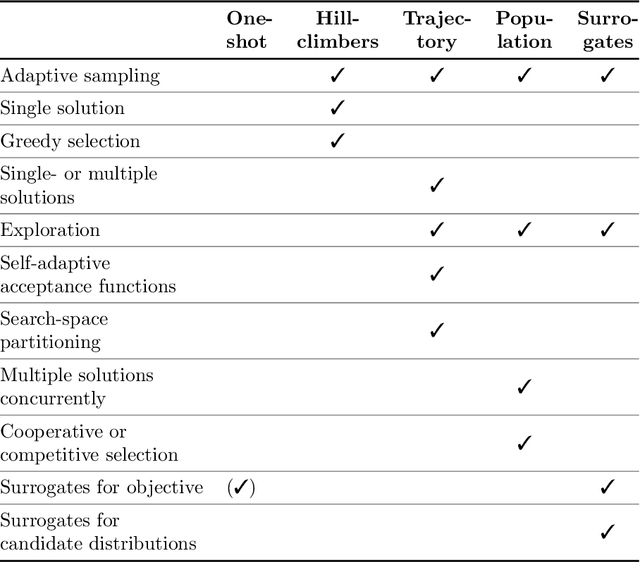
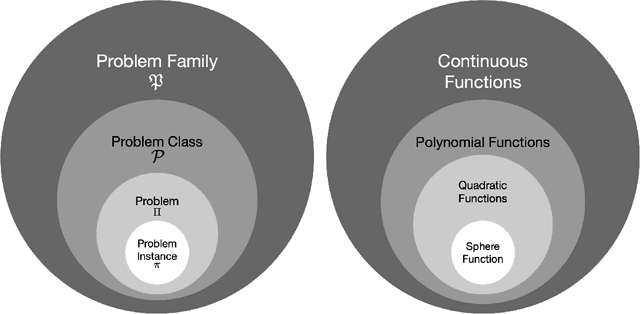

This survey compiles ideas and recommendations from more than a dozen researchers with different backgrounds and from different institutes around the world. Promoting best practice in benchmarking is its main goal. The article discusses eight essential topics in benchmarking: clearly stated goals, well-specified problems, suitable algorithms, adequate performance measures, thoughtful analysis, effective and efficient designs, comprehensible presentations, and guaranteed reproducibility. The final goal is to provide well-accepted guidelines (rules) that might be useful for authors and reviewers. As benchmarking in optimization is an active and evolving field of research this manuscript is meant to co-evolve over time by means of periodic updates.
EBIC: an evolutionary-based parallel biclustering algorithm for pattern discover
Jul 26, 2018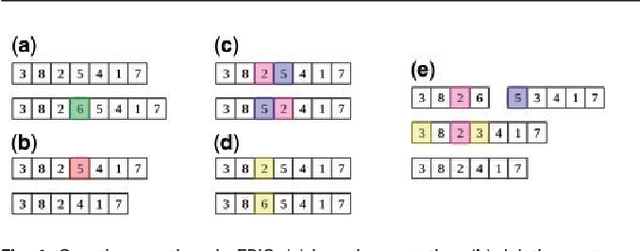
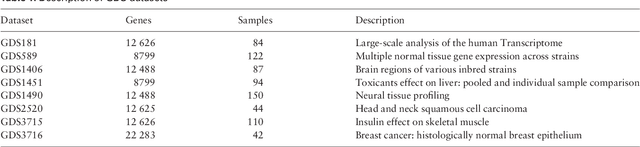
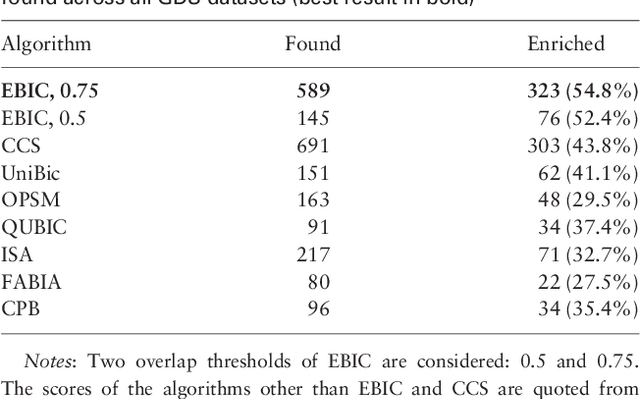
In this paper a novel biclustering algorithm based on artificial intelligence (AI) is introduced. The method called EBIC aims to detect biologically meaningful, order-preserving patterns in complex data. The proposed algorithm is probably the first one capable of discovering with accuracy exceeding 50% multiple complex patterns in real gene expression datasets. It is also one of the very few biclustering methods designed for parallel environments with multiple graphics processing units (GPUs). We demonstrate that EBIC outperforms state-of-the-art biclustering methods, in terms of recovery and relevance, on both synthetic and genetic datasets. EBIC also yields results over 12 times faster than the most accurate reference algorithms. The proposed algorithm is anticipated to be added to the repertoire of unsupervised machine learning algorithms for the analysis of datasets, including those from large-scale genomic studies.
EBIC: an open source software for high-dimensional and big data biclustering analyses
Jul 26, 2018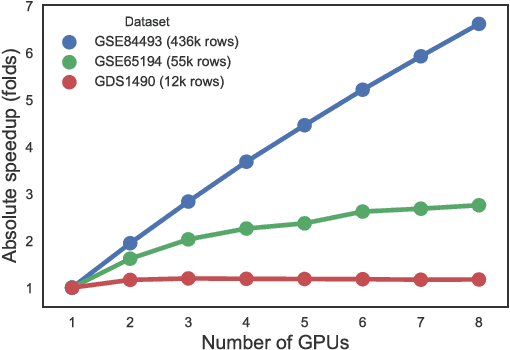

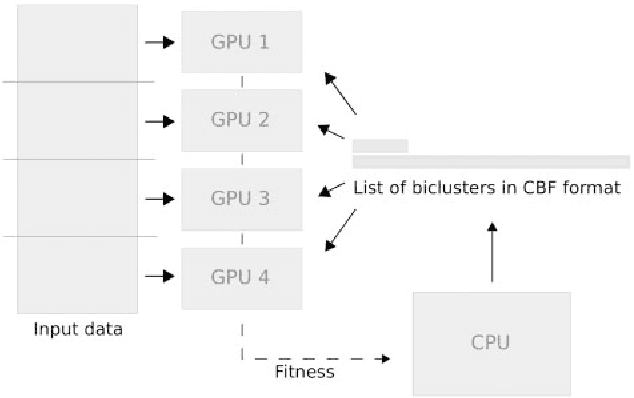
Motivation: In this paper we present the latest release of EBIC, a next-generation biclustering algorithm for mining genetic data. The major contribution of this paper is adding support for big data, making it possible to efficiently run large genomic data mining analyses. Additional enhancements include integration with R and Bioconductor and an option to remove influence of missing value on the final result. Results: EBIC was applied to datasets of different sizes, including a large DNA methylation dataset with 436,444 rows. For the largest dataset we observed over 6.6 fold speedup in computation time on a cluster of 8 GPUs compared to running the method on a single GPU. This proves high scalability of the algorithm. Availability: The latest version of EBIC could be downloaded from http://github.com/EpistasisLab/ebic . Installation and usage instructions are also available online.
Where are we now? A large benchmark study of recent symbolic regression methods
Jun 07, 2018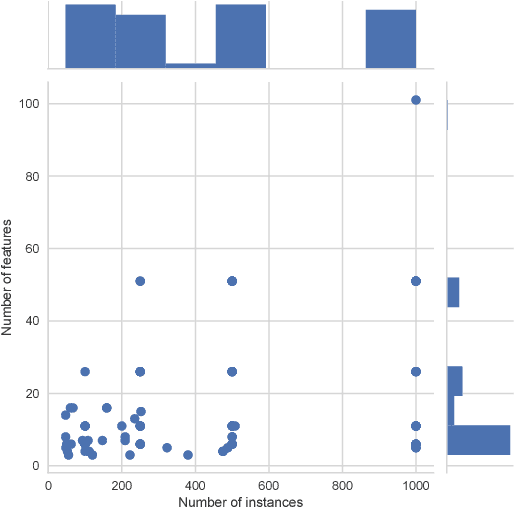
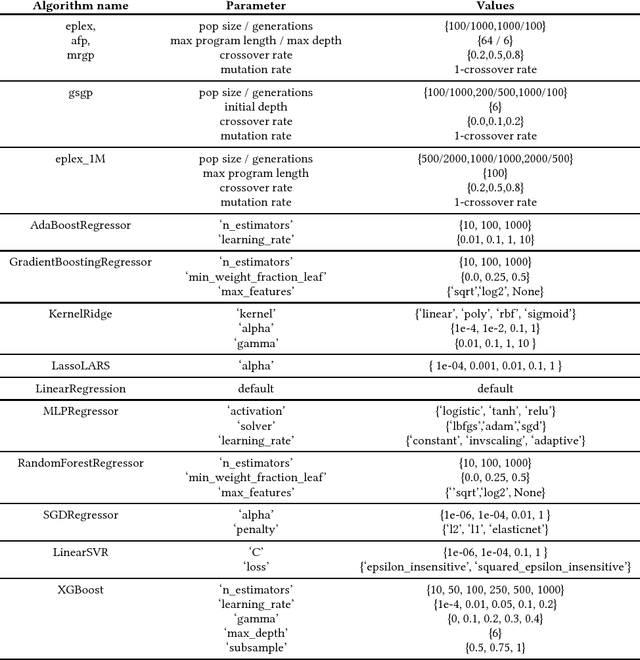
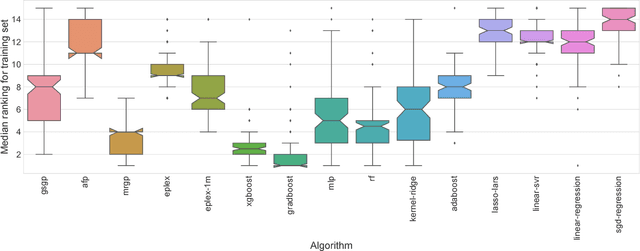

In this paper we provide a broad benchmarking of recent genetic programming approaches to symbolic regression in the context of state of the art machine learning approaches. We use a set of nearly 100 regression benchmark problems culled from open source repositories across the web. We conduct a rigorous benchmarking of four recent symbolic regression approaches as well as nine machine learning approaches from scikit-learn. The results suggest that symbolic regression performs strongly compared to state-of-the-art gradient boosting algorithms, although in terms of running times is among the slowest of the available methodologies. We discuss the results in detail and point to future research directions that may allow symbolic regression to gain wider adoption in the machine learning community.