 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Monosemantic Attribution Framework for Stable Interpretability in Clinical Neuroscience Large Language Models
Jan 25, 2026Interpretability remains a key challenge for deploying large language models (LLMs) in clinical settings such as Alzheimer's disease progression diagnosis, where early and trustworthy predictions are essential. Existing attribution methods exhibit high inter-method variability and unstable explanations due to the polysemantic nature of LLM representations, while mechanistic interpretability approaches lack direct alignment with model inputs and outputs and do not provide explicit importance scores. We introduce a unified interpretability framework that integrates attributional and mechanistic perspectives through monosemantic feature extraction. By constructing a monosemantic embedding space at the level of an LLM layer and optimizing the framework to explicitly reduce inter-method variability, our approach produces stable input-level importance scores and highlights salient features via a decompressed representation of the layer of interest, advancing the safe and trustworthy application of LLMs in cognitive health and neurodegenerative disease.
Unified Generative Latent Representation for Functional Brain Graphs
Nov 06, 2025Functional brain graphs are often characterized with separate graph-theoretic or spectral descriptors, overlooking how these properties covary and partially overlap across brains and conditions. We anticipate that dense, weighted functional connectivity graphs occupy a low-dimensional latent geometry along which both topological and spectral structures display graded variations. Here, we estimated this unified graph representation and enabled generation of dense functional brain graphs through a graph transformer autoencoder with latent diffusion, with spectral geometry providing an inductive bias to guide learning. This geometry-aware latent representation, although unsupervised, meaningfully separated working-memory states and decoded visual stimuli, with performance further enhanced by incorporating neural dynamics. From the diffusion modeled distribution, we were able to sample biologically plausible and structurally grounded synthetic dense graphs.
TourSynbio: A Multi-Modal Large Model and Agent Framework to Bridge Text and Protein Sequences for Protein Engineering
Aug 27, 2024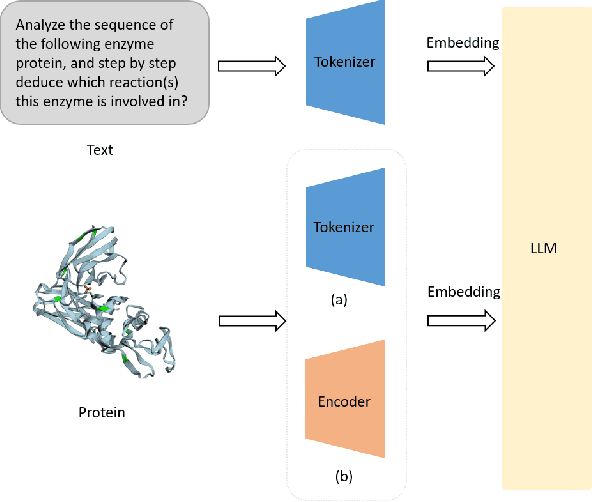
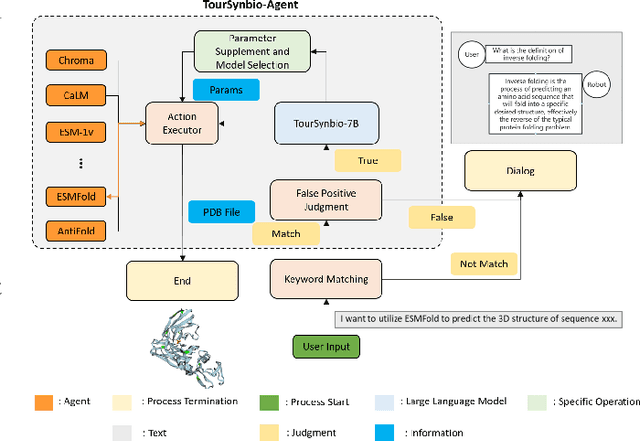
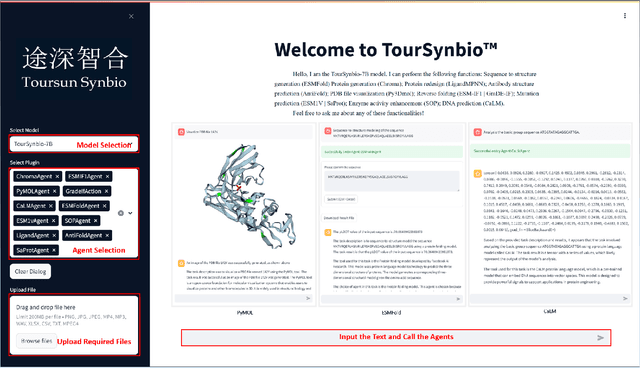
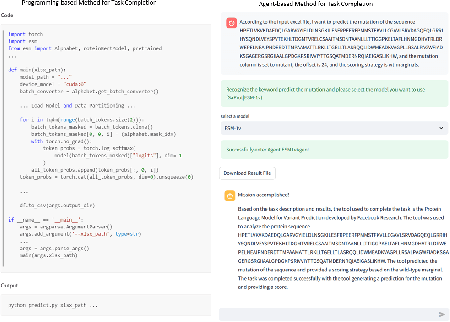
The structural similarities between protein sequences and natural languages have led to parallel advancements in deep learning across both domains. While large language models (LLMs) have achieved much progress in the domain of natural language processing, their potential in protein engineering remains largely unexplored. Previous approaches have equipped LLMs with protein understanding capabilities by incorporating external protein encoders, but this fails to fully leverage the inherent similarities between protein sequences and natural languages, resulting in sub-optimal performance and increased model complexity. To address this gap, we present TourSynbio-7B, the first multi-modal large model specifically designed for protein engineering tasks without external protein encoders. TourSynbio-7B demonstrates that LLMs can inherently learn to understand proteins as language. The model is post-trained and instruction fine-tuned on InternLM2-7B using ProteinLMDataset, a dataset comprising 17.46 billion tokens of text and protein sequence for self-supervised pretraining and 893K instructions for supervised fine-tuning. TourSynbio-7B outperforms GPT-4 on the ProteinLMBench, a benchmark of 944 manually verified multiple-choice questions, with 62.18% accuracy. Leveraging TourSynbio-7B's enhanced protein sequence understanding capability, we introduce TourSynbio-Agent, an innovative framework capable of performing various protein engineering tasks, including mutation analysis, inverse folding, protein folding, and visualization. TourSynbio-Agent integrates previously disconnected deep learning models in the protein engineering domain, offering a unified conversational user interface for improved usability. Finally, we demonstrate the efficacy of TourSynbio-7B and TourSynbio-Agent through two wet lab case studies on vanilla key enzyme modification and steroid compound catalysis.
A Fine-tuning Dataset and Benchmark for Large Language Models for Protein Understanding
Jun 08, 2024The parallels between protein sequences and natural language in their sequential structures have inspired the application of large language models (LLMs) to protein understanding. Despite the success of LLMs in NLP, their effectiveness in comprehending protein sequences remains an open question, largely due to the absence of datasets linking protein sequences to descriptive text. Researchers have then attempted to adapt LLMs for protein understanding by integrating a protein sequence encoder with a pre-trained LLM. However, this adaptation raises a fundamental question: "Can LLMs, originally designed for NLP, effectively comprehend protein sequences as a form of language?" Current datasets fall short in addressing this question due to the lack of a direct correlation between protein sequences and corresponding text descriptions, limiting the ability to train and evaluate LLMs for protein understanding effectively. To bridge this gap, we introduce ProteinLMDataset, a dataset specifically designed for further self-supervised pretraining and supervised fine-tuning (SFT) of LLMs to enhance their capability for protein sequence comprehension. Specifically, ProteinLMDataset includes 17.46 billion tokens for pretraining and 893,000 instructions for SFT. Additionally, we present ProteinLMBench, the first benchmark dataset consisting of 944 manually verified multiple-choice questions for assessing the protein understanding capabilities of LLMs. ProteinLMBench incorporates protein-related details and sequences in multiple languages, establishing a new standard for evaluating LLMs' abilities in protein comprehension. The large language model InternLM2-7B, pretrained and fine-tuned on the ProteinLMDataset, outperforms GPT-4 on ProteinLMBench, achieving the highest accuracy score. The dataset and the benchmark are available at https://huggingface.co/datasets/tsynbio/ProteinLMBench.
The Explanation Necessity for Healthcare AI
May 31, 2024


Explainability is often critical to the acceptable implementation of artificial intelligence (AI). Nowhere is this more important than healthcare where decision-making directly impacts patients and trust in AI systems is essential. This trust is often built on the explanations and interpretations the AI provides. Despite significant advancements in AI interpretability, there remains the need for clear guidelines on when and to what extent explanations are necessary in the medical context. We propose a novel categorization system with four distinct classes of explanation necessity, guiding the level of explanation required: patient or sample (local) level, cohort or dataset (global) level, or both levels. We introduce a mathematical formulation that distinguishes these categories and offers a practical framework for researchers to determine the necessity and depth of explanations required in medical AI applications. Three key factors are considered: the robustness of the evaluation protocol, the variability of expert observations, and the representation dimensionality of the application. In this perspective, we address the question: When does an AI medical application need to be explained, and at what level of detail?
Contrastive-Adversarial and Diffusion: Exploring pre-training and fine-tuning strategies for sulcal identification
May 29, 2024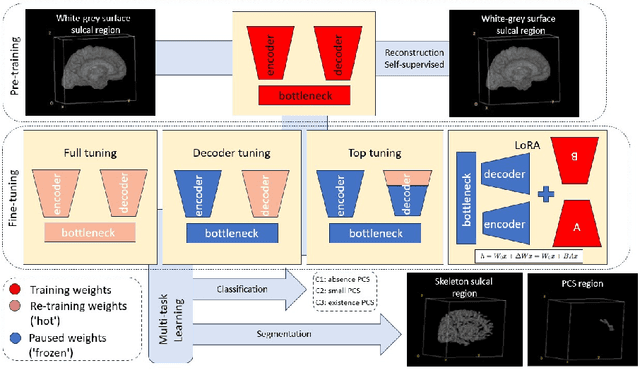
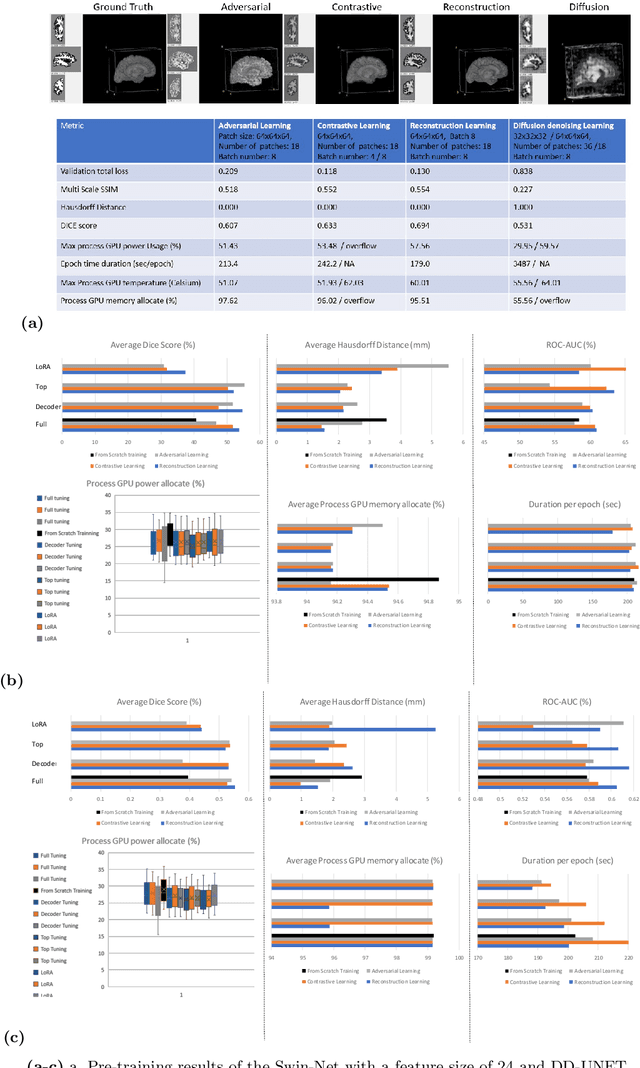
In the last decade, computer vision has witnessed the establishment of various training and learning approaches. Techniques like adversarial learning, contrastive learning, diffusion denoising learning, and ordinary reconstruction learning have become standard, representing state-of-the-art methods extensively employed for fully training or pre-training networks across various vision tasks. The exploration of fine-tuning approaches has emerged as a current focal point, addressing the need for efficient model tuning with reduced GPU memory usage and time costs while enhancing overall performance, as exemplified by methodologies like low-rank adaptation (LoRA). Key questions arise: which pre-training technique yields optimal results - adversarial, contrastive, reconstruction, or diffusion denoising? How does the performance of these approaches vary as the complexity of fine-tuning is adjusted? This study aims to elucidate the advantages of pre-training techniques and fine-tuning strategies to enhance the learning process of neural networks in independent identical distribution (IID) cohorts. We underscore the significance of fine-tuning by examining various cases, including full tuning, decoder tuning, top-level tuning, and fine-tuning of linear parameters using LoRA. Systematic summaries of model performance and efficiency are presented, leveraging metrics such as accuracy, time cost, and memory efficiency. To empirically demonstrate our findings, we focus on a multi-task segmentation-classification challenge involving the paracingulate sulcus (PCS) using different 3D Convolutional Neural Network (CNN) architectures by using the TOP-OSLO cohort comprising 596 subjects.
Solving the enigma: Deriving optimal explanations of deep networks
May 16, 2024The accelerated progress of artificial intelligence (AI) has popularized deep learning models across domains, yet their inherent opacity poses challenges, notably in critical fields like healthcare, medicine and the geosciences. Explainable AI (XAI) has emerged to shed light on these "black box" models, helping decipher their decision making process. Nevertheless, different XAI methods yield highly different explanations. This inter-method variability increases uncertainty and lowers trust in deep networks' predictions. In this study, for the first time, we propose a novel framework designed to enhance the explainability of deep networks, by maximizing both the accuracy and the comprehensibility of the explanations. Our framework integrates various explanations from established XAI methods and employs a non-linear "explanation optimizer" to construct a unique and optimal explanation. Through experiments on multi-class and binary classification tasks in 2D object and 3D neuroscience imaging, we validate the efficacy of our approach. Our explanation optimizer achieved superior faithfulness scores, averaging 155% and 63% higher than the best performing XAI method in the 3D and 2D applications, respectively. Additionally, our approach yielded lower complexity, increasing comprehensibility. Our results suggest that optimal explanations based on specific criteria are derivable and address the issue of inter-method variability in the current XAI literature.
Hunting imaging biomarkers in pulmonary fibrosis: Benchmarks of the AIIB23 challenge
Dec 21, 2023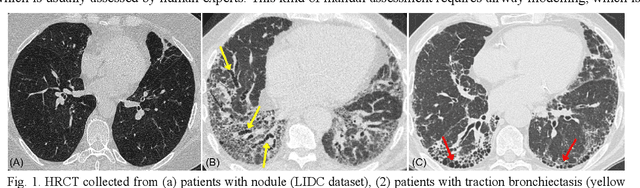
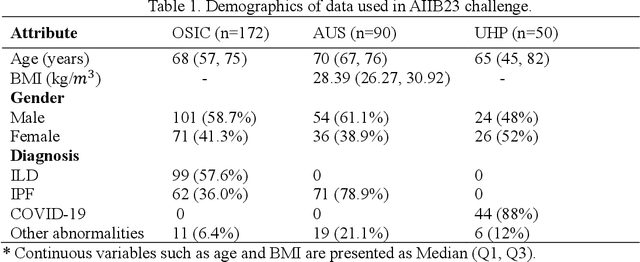
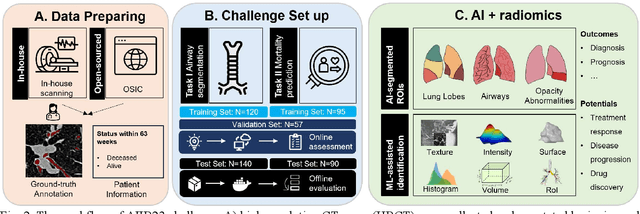
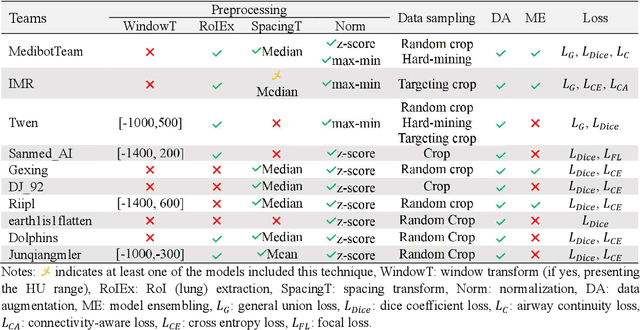
Airway-related quantitative imaging biomarkers are crucial for examination, diagnosis, and prognosis in pulmonary diseases. However, the manual delineation of airway trees remains prohibitively time-consuming. While significant efforts have been made towards enhancing airway modelling, current public-available datasets concentrate on lung diseases with moderate morphological variations. The intricate honeycombing patterns present in the lung tissues of fibrotic lung disease patients exacerbate the challenges, often leading to various prediction errors. To address this issue, the 'Airway-Informed Quantitative CT Imaging Biomarker for Fibrotic Lung Disease 2023' (AIIB23) competition was organized in conjunction with the official 2023 International Conference on Medical Image Computing and Computer Assisted Intervention (MICCAI). The airway structures were meticulously annotated by three experienced radiologists. Competitors were encouraged to develop automatic airway segmentation models with high robustness and generalization abilities, followed by exploring the most correlated QIB of mortality prediction. A training set of 120 high-resolution computerised tomography (HRCT) scans were publicly released with expert annotations and mortality status. The online validation set incorporated 52 HRCT scans from patients with fibrotic lung disease and the offline test set included 140 cases from fibrosis and COVID-19 patients. The results have shown that the capacity of extracting airway trees from patients with fibrotic lung disease could be enhanced by introducing voxel-wise weighted general union loss and continuity loss. In addition to the competitive image biomarkers for prognosis, a strong airway-derived biomarker (Hazard ratio>1.5, p<0.0001) was revealed for survival prognostication compared with existing clinical measurements, clinician assessment and AI-based biomarkers.
A 3D explainability framework to uncover learning patterns and crucial sub-regions in variable sulci recognition
Sep 02, 2023

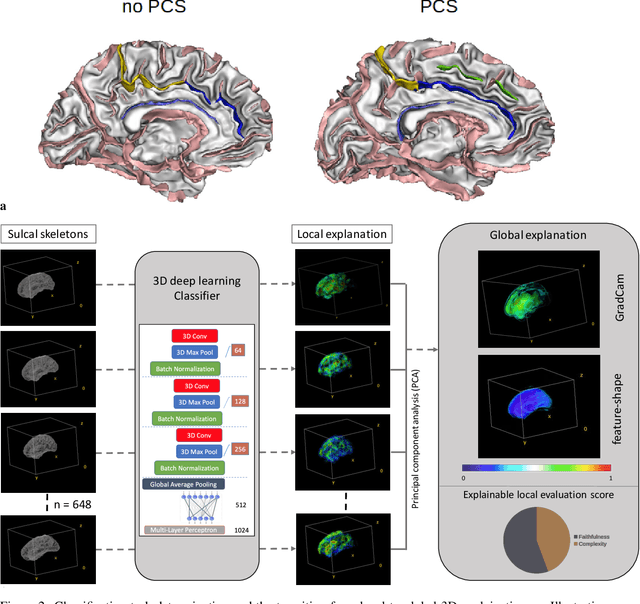

Precisely identifying sulcal features in brain MRI is made challenging by the variability of brain folding. This research introduces an innovative 3D explainability frame-work that validates outputs from deep learning networks in their ability to detect the paracingulate sulcus, an anatomical feature that may or may not be present on the frontal medial surface of the human brain. This study trained and tested two networks, amalgamating local explainability techniques GradCam and SHAP with a dimensionality reduction method. The explainability framework provided both localized and global explanations, along with accuracy of classification results, revealing pertinent sub-regions contributing to the decision process through a post-fusion transformation of explanatory and statistical features. Leveraging the TOP-OSLO dataset of MRI acquired from patients with schizophrenia, greater accuracies of paracingulate sulcus detection (presence or absence) were found in the left compared to right hemispheres with distinct, but extensive sub-regions contributing to each classification outcome. The study also inadvertently highlighted the critical role of an unbiased annotation protocol in maintaining network performance fairness. Our proposed method not only offers automated, impartial annotations of a variable sulcus but also provides insights into the broader anatomical variations associated with its presence throughout the brain. The adoption of this methodology holds promise for instigating further explorations and inquiries in the field of neuroscience.
A novel framework employing deep multi-attention channels network for the autonomous detection of metastasizing cells through fluorescence microscopy
Sep 02, 2023We developed a transparent computational large-scale imaging-based framework that can distinguish between normal and metastasizing human cells. The method relies on fluorescence microscopy images showing the spatial organization of actin and vimentin filaments in normal and metastasizing single cells, using a combination of multi-attention channels network and global explainable techniques. We test a classification between normal cells (Bj primary fibroblast), and their isogenically matched, transformed and invasive counterpart (BjTertSV40TRasV12). Manual annotation is not trivial to automate due to the intricacy of the biologically relevant features. In this research, we utilized established deep learning networks and our new multi-attention channel architecture. To increase the interpretability of the network - crucial for this application area - we developed an interpretable global explainable approach correlating the weighted geometric mean of the total cell images and their local GradCam scores. The significant results from our analysis unprecedently allowed a more detailed, and biologically relevant understanding of the cytoskeletal changes that accompany oncogenic transformation of normal to invasive and metastasizing cells. We also paved the way for a possible spatial micrometre-level biomarker for future development of diagnostic tools against metastasis (spatial distribution of vimentin).