 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Frequency-Aware Self-Supervised Learning for Ultra-Wide-Field Image Enhancement
Aug 27, 2025Ultra-Wide-Field (UWF) retinal imaging has revolutionized retinal diagnostics by providing a comprehensive view of the retina. However, it often suffers from quality-degrading factors such as blurring and uneven illumination, which obscure fine details and mask pathological information. While numerous retinal image enhancement methods have been proposed for other fundus imageries, they often fail to address the unique requirements in UWF, particularly the need to preserve pathological details. In this paper, we propose a novel frequency-aware self-supervised learning method for UWF image enhancement. It incorporates frequency-decoupled image deblurring and Retinex-guided illumination compensation modules. An asymmetric channel integration operation is introduced in the former module, so as to combine global and local views by leveraging high- and low-frequency information, ensuring the preservation of fine and broader structural details. In addition, a color preservation unit is proposed in the latter Retinex-based module, to provide multi-scale spatial and frequency information, enabling accurate illumination estimation and correction. Experimental results demonstrate that the proposed work not only enhances visualization quality but also improves disease diagnosis performance by restoring and correcting fine local details and uneven intensity. To the best of our knowledge, this work is the first attempt for UWF image enhancement, offering a robust and clinically valuable tool for improving retinal disease management.
A Survey for Large Language Models in Biomedicine
Aug 29, 2024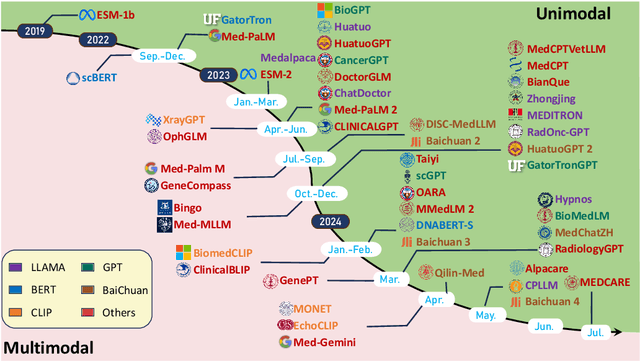
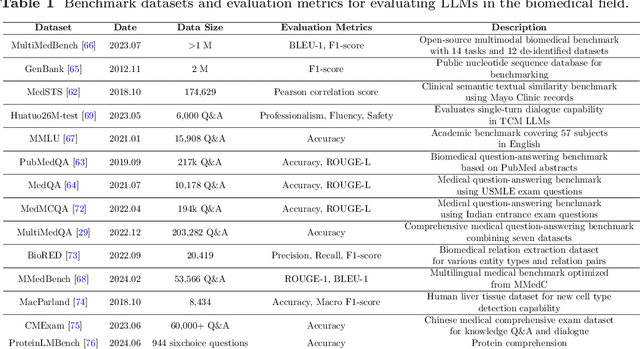
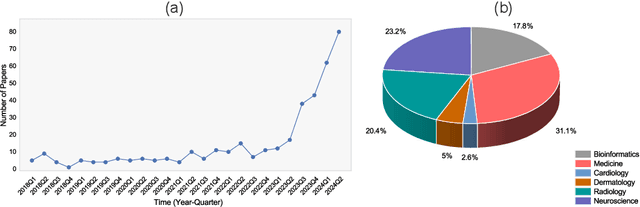
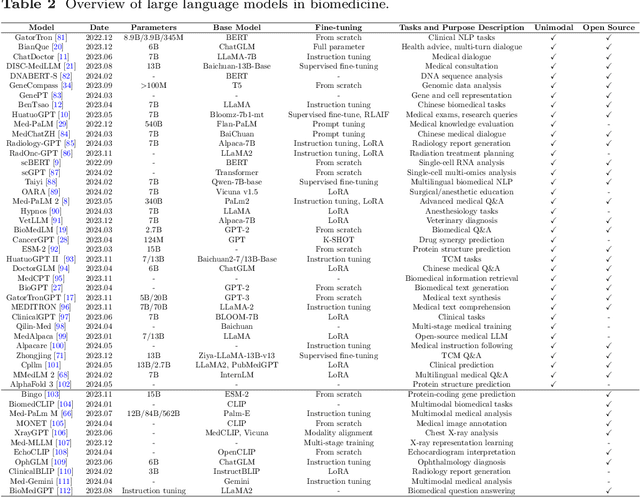
Recent breakthroughs in large language models (LLMs) offer unprecedented natural language understanding and generation capabilities. However, existing surveys on LLMs in biomedicine often focus on specific applications or model architectures, lacking a comprehensive analysis that integrates the latest advancements across various biomedical domains. This review, based on an analysis of 484 publications sourced from databases including PubMed, Web of Science, and arXiv, provides an in-depth examination of the current landscape, applications, challenges, and prospects of LLMs in biomedicine, distinguishing itself by focusing on the practical implications of these models in real-world biomedical contexts. Firstly, we explore the capabilities of LLMs in zero-shot learning across a broad spectrum of biomedical tasks, including diagnostic assistance, drug discovery, and personalized medicine, among others, with insights drawn from 137 key studies. Then, we discuss adaptation strategies of LLMs, including fine-tuning methods for both uni-modal and multi-modal LLMs to enhance their performance in specialized biomedical contexts where zero-shot fails to achieve, such as medical question answering and efficient processing of biomedical literature. Finally, we discuss the challenges that LLMs face in the biomedicine domain including data privacy concerns, limited model interpretability, issues with dataset quality, and ethics due to the sensitive nature of biomedical data, the need for highly reliable model outputs, and the ethical implications of deploying AI in healthcare. To address these challenges, we also identify future research directions of LLM in biomedicine including federated learning methods to preserve data privacy and integrating explainable AI methodologies to enhance the transparency of LLMs.
TourSynbio: A Multi-Modal Large Model and Agent Framework to Bridge Text and Protein Sequences for Protein Engineering
Aug 27, 2024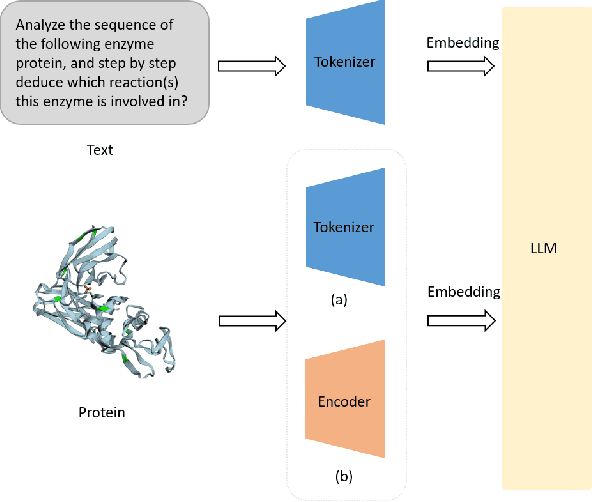
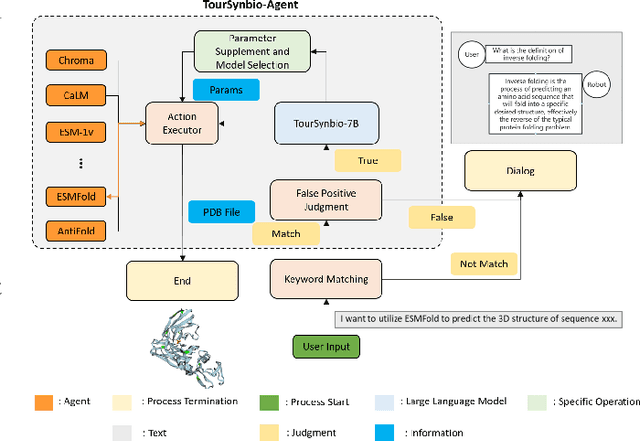
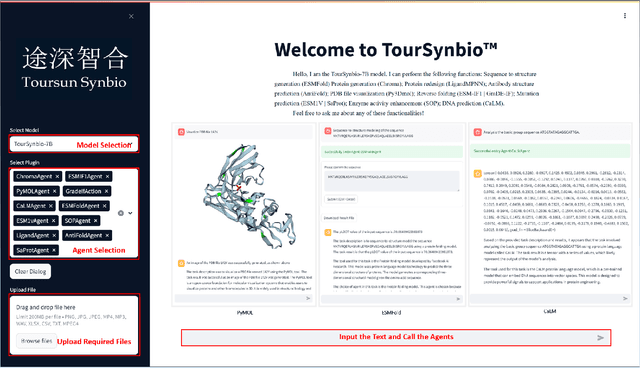
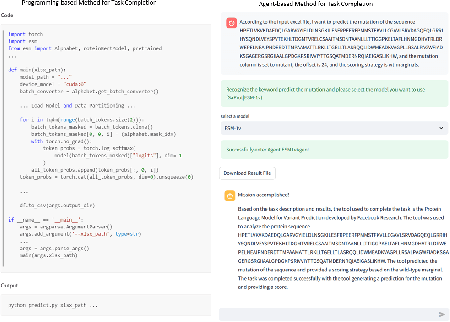
The structural similarities between protein sequences and natural languages have led to parallel advancements in deep learning across both domains. While large language models (LLMs) have achieved much progress in the domain of natural language processing, their potential in protein engineering remains largely unexplored. Previous approaches have equipped LLMs with protein understanding capabilities by incorporating external protein encoders, but this fails to fully leverage the inherent similarities between protein sequences and natural languages, resulting in sub-optimal performance and increased model complexity. To address this gap, we present TourSynbio-7B, the first multi-modal large model specifically designed for protein engineering tasks without external protein encoders. TourSynbio-7B demonstrates that LLMs can inherently learn to understand proteins as language. The model is post-trained and instruction fine-tuned on InternLM2-7B using ProteinLMDataset, a dataset comprising 17.46 billion tokens of text and protein sequence for self-supervised pretraining and 893K instructions for supervised fine-tuning. TourSynbio-7B outperforms GPT-4 on the ProteinLMBench, a benchmark of 944 manually verified multiple-choice questions, with 62.18% accuracy. Leveraging TourSynbio-7B's enhanced protein sequence understanding capability, we introduce TourSynbio-Agent, an innovative framework capable of performing various protein engineering tasks, including mutation analysis, inverse folding, protein folding, and visualization. TourSynbio-Agent integrates previously disconnected deep learning models in the protein engineering domain, offering a unified conversational user interface for improved usability. Finally, we demonstrate the efficacy of TourSynbio-7B and TourSynbio-Agent through two wet lab case studies on vanilla key enzyme modification and steroid compound catalysis.
A Fine-tuning Dataset and Benchmark for Large Language Models for Protein Understanding
Jun 08, 2024The parallels between protein sequences and natural language in their sequential structures have inspired the application of large language models (LLMs) to protein understanding. Despite the success of LLMs in NLP, their effectiveness in comprehending protein sequences remains an open question, largely due to the absence of datasets linking protein sequences to descriptive text. Researchers have then attempted to adapt LLMs for protein understanding by integrating a protein sequence encoder with a pre-trained LLM. However, this adaptation raises a fundamental question: "Can LLMs, originally designed for NLP, effectively comprehend protein sequences as a form of language?" Current datasets fall short in addressing this question due to the lack of a direct correlation between protein sequences and corresponding text descriptions, limiting the ability to train and evaluate LLMs for protein understanding effectively. To bridge this gap, we introduce ProteinLMDataset, a dataset specifically designed for further self-supervised pretraining and supervised fine-tuning (SFT) of LLMs to enhance their capability for protein sequence comprehension. Specifically, ProteinLMDataset includes 17.46 billion tokens for pretraining and 893,000 instructions for SFT. Additionally, we present ProteinLMBench, the first benchmark dataset consisting of 944 manually verified multiple-choice questions for assessing the protein understanding capabilities of LLMs. ProteinLMBench incorporates protein-related details and sequences in multiple languages, establishing a new standard for evaluating LLMs' abilities in protein comprehension. The large language model InternLM2-7B, pretrained and fine-tuned on the ProteinLMDataset, outperforms GPT-4 on ProteinLMBench, achieving the highest accuracy score. The dataset and the benchmark are available at https://huggingface.co/datasets/tsynbio/ProteinLMBench.
DeepMpMRI: Tensor-decomposition Regularized Learning for Fast and High-Fidelity Multi-Parametric Microstructural MR Imaging
May 06, 2024Deep learning has emerged as a promising approach for learning the nonlinear mapping between diffusion-weighted MR images and tissue parameters, which enables automatic and deep understanding of the brain microstructures. However, the efficiency and accuracy in the multi-parametric estimations are still limited since previous studies tend to estimate multi-parametric maps with dense sampling and isolated signal modeling. This paper proposes DeepMpMRI, a unified framework for fast and high-fidelity multi-parametric estimation from various diffusion models using sparsely sampled q-space data. DeepMpMRI is equipped with a newly designed tensor-decomposition-based regularizer to effectively capture fine details by exploiting the correlation across parameters. In addition, we introduce a Nesterov-based adaptive learning algorithm that optimizes the regularization parameter dynamically to enhance the performance. DeepMpMRI is an extendable framework capable of incorporating flexible network architecture. Experimental results demonstrate the superiority of our approach over 5 state-of-the-art methods in simultaneously estimating multi-parametric maps for various diffusion models with fine-grained details both quantitatively and qualitatively, achieving 4.5 - 22.5$\times$ acceleration compared to the dense sampling of a total of 270 diffusion gradients.