 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultiscale guidance of AlphaFold3 with heterogeneous cryo-EM data
Jun 04, 2025Protein structure prediction models are now capable of generating accurate 3D structural hypotheses from sequence alone. However, they routinely fail to capture the conformational diversity of dynamic biomolecular complexes, often requiring heuristic MSA subsampling approaches for generating alternative states. In parallel, cryo-electron microscopy (cryo-EM) has emerged as a powerful tool for imaging near-native structural heterogeneity, but is challenged by arduous pipelines to go from raw experimental data to atomic models. Here, we bridge the gap between these modalities, combining cryo-EM density maps with the rich sequence and biophysical priors learned by protein structure prediction models. Our method, CryoBoltz, guides the sampling trajectory of a pretrained protein structure prediction model using both global and local structural constraints derived from density maps, driving predictions towards conformational states consistent with the experimental data. We demonstrate that this flexible yet powerful inference-time approach allows us to build atomic models into heterogeneous cryo-EM maps across a variety of dynamic biomolecular systems including transporters and antibodies.
Dual Ascent Diffusion for Inverse Problems
May 23, 2025Ill-posed inverse problems are fundamental in many domains, ranging from astrophysics to medical imaging. Emerging diffusion models provide a powerful prior for solving these problems. Existing maximum-a-posteriori (MAP) or posterior sampling approaches, however, rely on different computational approximations, leading to inaccurate or suboptimal samples. To address this issue, we introduce a new approach to solving MAP problems with diffusion model priors using a dual ascent optimization framework. Our framework achieves better image quality as measured by various metrics for image restoration problems, it is more robust to high levels of measurement noise, it is faster, and it estimates solutions that represent the observations more faithfully than the state of the art.
Mixture of neural fields for heterogeneous reconstruction in cryo-EM
Dec 12, 2024



Cryo-electron microscopy (cryo-EM) is an experimental technique for protein structure determination that images an ensemble of macromolecules in near-physiological contexts. While recent advances enable the reconstruction of dynamic conformations of a single biomolecular complex, current methods do not adequately model samples with mixed conformational and compositional heterogeneity. In particular, datasets containing mixtures of multiple proteins require the joint inference of structure, pose, compositional class, and conformational states for 3D reconstruction. Here, we present Hydra, an approach that models both conformational and compositional heterogeneity fully ab initio by parameterizing structures as arising from one of K neural fields. We employ a new likelihood-based loss function and demonstrate the effectiveness of our approach on synthetic datasets composed of mixtures of proteins with large degrees of conformational variability. We additionally demonstrate Hydra on an experimental dataset of a cellular lysate containing a mixture of different protein complexes. Hydra expands the expressivity of heterogeneous reconstruction methods and thus broadens the scope of cryo-EM to increasingly complex samples.
Solving Inverse Problems in Protein Space Using Diffusion-Based Priors
Jun 06, 2024The interaction of a protein with its environment can be understood and controlled via its 3D structure. Experimental methods for protein structure determination, such as X-ray crystallography or cryogenic electron microscopy, shed light on biological processes but introduce challenging inverse problems. Learning-based approaches have emerged as accurate and efficient methods to solve these inverse problems for 3D structure determination, but are specialized for a predefined type of measurement. Here, we introduce a versatile framework to turn raw biophysical measurements of varying types into 3D atomic models. Our method combines a physics-based forward model of the measurement process with a pretrained generative model providing a task-agnostic, data-driven prior. Our method outperforms posterior sampling baselines on both linear and non-linear inverse problems. In particular, it is the first diffusion-based method for refining atomic models from cryo-EM density maps.
Scalable 3D Reconstruction From Single Particle X-Ray Diffraction Images Based on Online Machine Learning
Dec 22, 2023X-ray free-electron lasers (XFELs) offer unique capabilities for measuring the structure and dynamics of biomolecules, helping us understand the basic building blocks of life. Notably, high-repetition-rate XFELs enable single particle imaging (X-ray SPI) where individual, weakly scattering biomolecules are imaged under near-physiological conditions with the opportunity to access fleeting states that cannot be captured in cryogenic or crystallized conditions. Existing X-ray SPI reconstruction algorithms, which estimate the unknown orientation of a particle in each captured image as well as its shared 3D structure, are inadequate in handling the massive datasets generated by these emerging XFELs. Here, we introduce X-RAI, an online reconstruction framework that estimates the structure of a 3D macromolecule from large X-ray SPI datasets. X-RAI consists of a convolutional encoder, which amortizes pose estimation over large datasets, as well as a physics-based decoder, which employs an implicit neural representation to enable high-quality 3D reconstruction in an end-to-end, self-supervised manner. We demonstrate that X-RAI achieves state-of-the-art performance for small-scale datasets in simulation and challenging experimental settings and demonstrate its unprecedented ability to process large datasets containing millions of diffraction images in an online fashion. These abilities signify a paradigm shift in X-ray SPI towards real-time capture and reconstruction.
Reconstructing Heterogeneous Cryo-EM Molecular Structures by Decomposing Them into Polymer Chains
Jun 12, 2023



Cryogenic electron microscopy (cryo-EM) has transformed structural biology by allowing to reconstruct 3D biomolecular structures up to near-atomic resolution. However, the 3D reconstruction process remains challenging, as the 3D structures may exhibit substantial shape variations, while the 2D image acquisition suffers from a low signal-to-noise ratio, requiring to acquire very large datasets that are time-consuming to process. Current reconstruction methods are precise but computationally expensive, or faster but lack a physically-plausible model of large molecular shape variations. To fill this gap, we propose CryoChains that encodes large deformations of biomolecules via rigid body transformation of their polymer instances (chains), while representing their finer shape variations with the normal mode analysis framework of biophysics. Our synthetic data experiments on the human $\text{GABA}_{\text{B}}$ and heat shock protein show that CryoChains gives a biophysically-grounded quantification of the heterogeneous conformations of biomolecules, while reconstructing their 3D molecular structures at an improved resolution compared to the current fastest, interpretable deep learning method.
Generative Novel View Synthesis with 3D-Aware Diffusion Models
Apr 05, 2023



We present a diffusion-based model for 3D-aware generative novel view synthesis from as few as a single input image. Our model samples from the distribution of possible renderings consistent with the input and, even in the presence of ambiguity, is capable of rendering diverse and plausible novel views. To achieve this, our method makes use of existing 2D diffusion backbones but, crucially, incorporates geometry priors in the form of a 3D feature volume. This latent feature field captures the distribution over possible scene representations and improves our method's ability to generate view-consistent novel renderings. In addition to generating novel views, our method has the ability to autoregressively synthesize 3D-consistent sequences. We demonstrate state-of-the-art results on synthetic renderings and room-scale scenes; we also show compelling results for challenging, real-world objects.
MELON: NeRF with Unposed Images Using Equivalence Class Estimation
Mar 14, 2023Neural radiance fields enable novel-view synthesis and scene reconstruction with photorealistic quality from a few images, but require known and accurate camera poses. Conventional pose estimation algorithms fail on smooth or self-similar scenes, while methods performing inverse rendering from unposed views require a rough initialization of the camera orientations. The main difficulty of pose estimation lies in real-life objects being almost invariant under certain transformations, making the photometric distance between rendered views non-convex with respect to the camera parameters. Using an equivalence relation that matches the distribution of local minima in camera space, we reduce this space to its quotient set, in which pose estimation becomes a more convex problem. Using a neural-network to regularize pose estimation, we demonstrate that our method - MELON - can reconstruct a neural radiance field from unposed images with state-of-the-art accuracy while requiring ten times fewer views than adversarial approaches.
Amortized Inference for Heterogeneous Reconstruction in Cryo-EM
Oct 13, 2022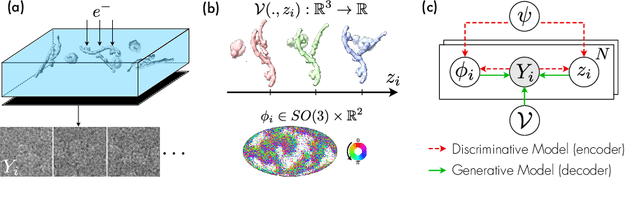
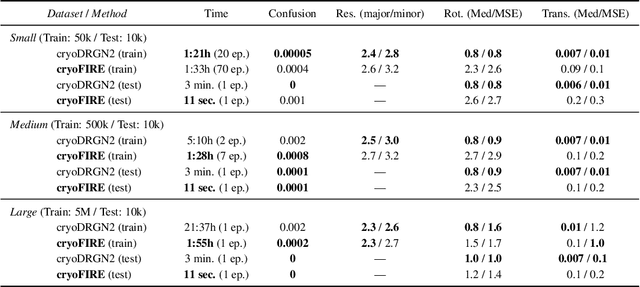
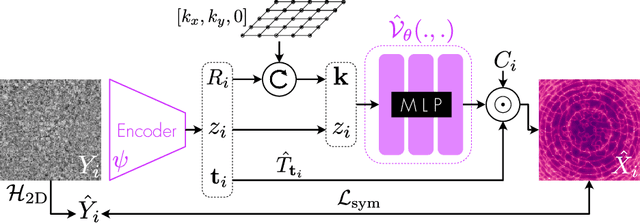

Cryo-electron microscopy (cryo-EM) is an imaging modality that provides unique insights into the dynamics of proteins and other building blocks of life. The algorithmic challenge of jointly estimating the poses, 3D structure, and conformational heterogeneity of a biomolecule from millions of noisy and randomly oriented 2D projections in a computationally efficient manner, however, remains unsolved. Our method, cryoFIRE, performs ab initio heterogeneous reconstruction with unknown poses in an amortized framework, thereby avoiding the computationally expensive step of pose search while enabling the analysis of conformational heterogeneity. Poses and conformation are jointly estimated by an encoder while a physics-based decoder aggregates the images into an implicit neural representation of the conformational space. We show that our method can provide one order of magnitude speedup on datasets containing millions of images without any loss of accuracy. We validate that the joint estimation of poses and conformations can be amortized over the size of the dataset. For the first time, we prove that an amortized method can extract interpretable dynamic information from experimental datasets.
Heterogeneous reconstruction of deformable atomic models in Cryo-EM
Sep 29, 2022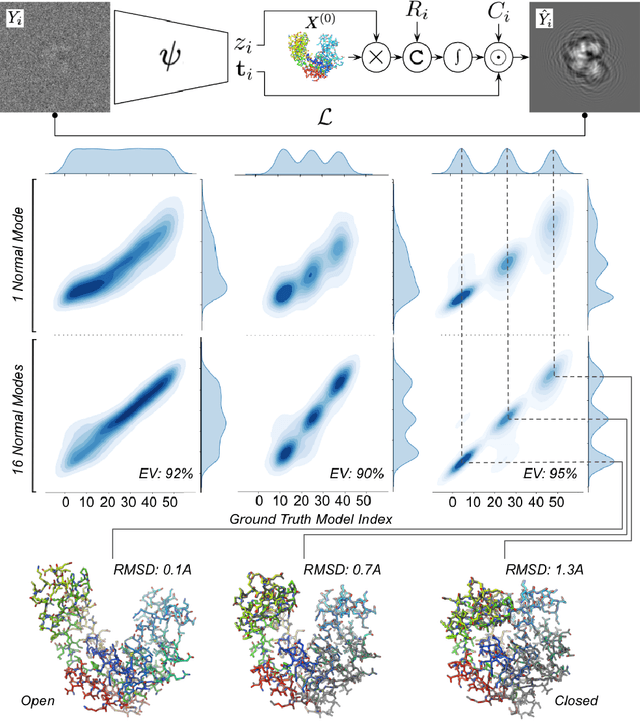
Cryogenic electron microscopy (cryo-EM) provides a unique opportunity to study the structural heterogeneity of biomolecules. Being able to explain this heterogeneity with atomic models would help our understanding of their functional mechanisms but the size and ruggedness of the structural space (the space of atomic 3D cartesian coordinates) presents an immense challenge. Here, we describe a heterogeneous reconstruction method based on an atomistic representation whose deformation is reduced to a handful of collective motions through normal mode analysis. Our implementation uses an autoencoder. The encoder jointly estimates the amplitude of motion along the normal modes and the 2D shift between the center of the image and the center of the molecule . The physics-based decoder aggregates a representation of the heterogeneity readily interpretable at the atomic level. We illustrate our method on 3 synthetic datasets corresponding to different distributions along a simulated trajectory of adenylate kinase transitioning from its open to its closed structures. We show for each distribution that our approach is able to recapitulate the intermediate atomic models with atomic-level accuracy.