 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReconstructing Heterogeneous Cryo-EM Molecular Structures by Decomposing Them into Polymer Chains
Jun 12, 2023



Cryogenic electron microscopy (cryo-EM) has transformed structural biology by allowing to reconstruct 3D biomolecular structures up to near-atomic resolution. However, the 3D reconstruction process remains challenging, as the 3D structures may exhibit substantial shape variations, while the 2D image acquisition suffers from a low signal-to-noise ratio, requiring to acquire very large datasets that are time-consuming to process. Current reconstruction methods are precise but computationally expensive, or faster but lack a physically-plausible model of large molecular shape variations. To fill this gap, we propose CryoChains that encodes large deformations of biomolecules via rigid body transformation of their polymer instances (chains), while representing their finer shape variations with the normal mode analysis framework of biophysics. Our synthetic data experiments on the human $\text{GABA}_{\text{B}}$ and heat shock protein show that CryoChains gives a biophysically-grounded quantification of the heterogeneous conformations of biomolecules, while reconstructing their 3D molecular structures at an improved resolution compared to the current fastest, interpretable deep learning method.
Amortized Inference for Heterogeneous Reconstruction in Cryo-EM
Oct 13, 2022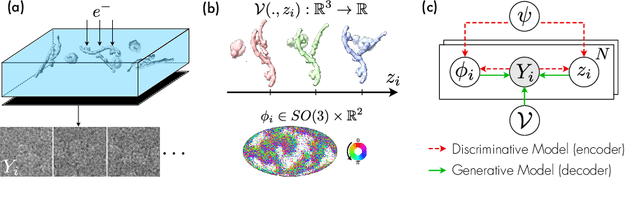
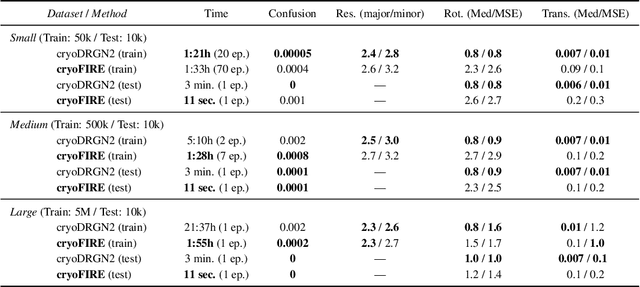
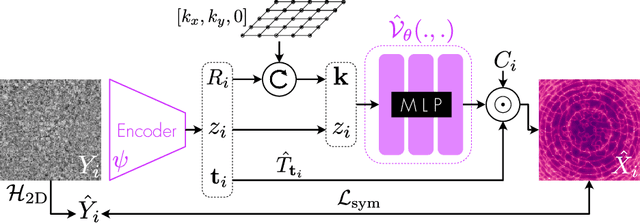

Cryo-electron microscopy (cryo-EM) is an imaging modality that provides unique insights into the dynamics of proteins and other building blocks of life. The algorithmic challenge of jointly estimating the poses, 3D structure, and conformational heterogeneity of a biomolecule from millions of noisy and randomly oriented 2D projections in a computationally efficient manner, however, remains unsolved. Our method, cryoFIRE, performs ab initio heterogeneous reconstruction with unknown poses in an amortized framework, thereby avoiding the computationally expensive step of pose search while enabling the analysis of conformational heterogeneity. Poses and conformation are jointly estimated by an encoder while a physics-based decoder aggregates the images into an implicit neural representation of the conformational space. We show that our method can provide one order of magnitude speedup on datasets containing millions of images without any loss of accuracy. We validate that the joint estimation of poses and conformations can be amortized over the size of the dataset. For the first time, we prove that an amortized method can extract interpretable dynamic information from experimental datasets.
Towards Trustworthy Automatic Diagnosis Systems by Emulating Doctors' Reasoning with Deep Reinforcement Learning
Oct 13, 2022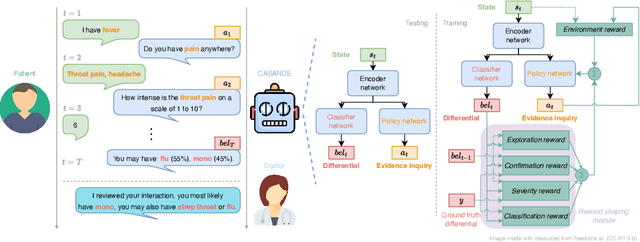
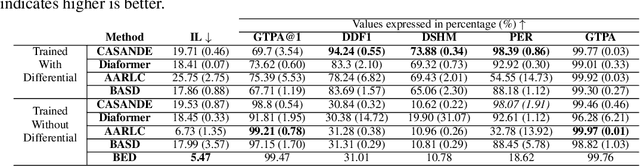
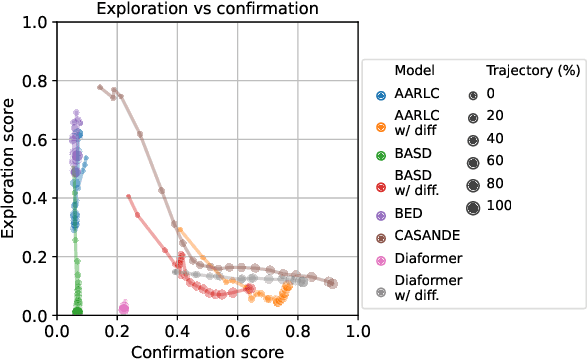

The automation of the medical evidence acquisition and diagnosis process has recently attracted increasing attention in order to reduce the workload of doctors and democratize access to medical care. However, most works proposed in the machine learning literature focus solely on improving the prediction accuracy of a patient's pathology. We argue that this objective is insufficient to ensure doctors' acceptability of such systems. In their initial interaction with patients, doctors do not only focus on identifying the pathology a patient is suffering from; they instead generate a differential diagnosis (in the form of a short list of plausible diseases) because the medical evidence collected from patients is often insufficient to establish a final diagnosis. Moreover, doctors explicitly explore severe pathologies before potentially ruling them out from the differential, especially in acute care settings. Finally, for doctors to trust a system's recommendations, they need to understand how the gathered evidences led to the predicted diseases. In particular, interactions between a system and a patient need to emulate the reasoning of doctors. We therefore propose to model the evidence acquisition and automatic diagnosis tasks using a deep reinforcement learning framework that considers three essential aspects of a doctor's reasoning, namely generating a differential diagnosis using an exploration-confirmation approach while prioritizing severe pathologies. We propose metrics for evaluating interaction quality based on these three aspects. We show that our approach performs better than existing models while maintaining competitive pathology prediction accuracy.
Heterogeneous reconstruction of deformable atomic models in Cryo-EM
Sep 29, 2022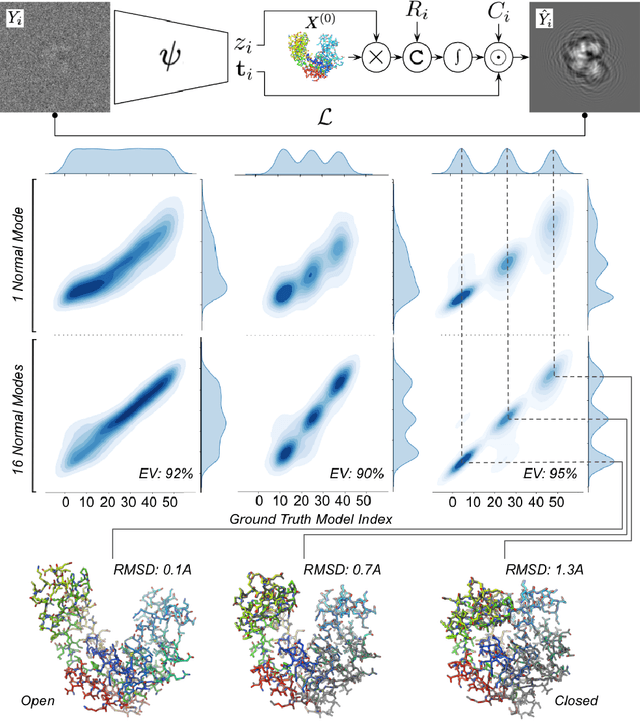
Cryogenic electron microscopy (cryo-EM) provides a unique opportunity to study the structural heterogeneity of biomolecules. Being able to explain this heterogeneity with atomic models would help our understanding of their functional mechanisms but the size and ruggedness of the structural space (the space of atomic 3D cartesian coordinates) presents an immense challenge. Here, we describe a heterogeneous reconstruction method based on an atomistic representation whose deformation is reduced to a handful of collective motions through normal mode analysis. Our implementation uses an autoencoder. The encoder jointly estimates the amplitude of motion along the normal modes and the 2D shift between the center of the image and the center of the molecule . The physics-based decoder aggregates a representation of the heterogeneity readily interpretable at the atomic level. We illustrate our method on 3 synthetic datasets corresponding to different distributions along a simulated trajectory of adenylate kinase transitioning from its open to its closed structures. We show for each distribution that our approach is able to recapitulate the intermediate atomic models with atomic-level accuracy.
CryoAI: Amortized Inference of Poses for Ab Initio Reconstruction of 3D Molecular Volumes from Real Cryo-EM Images
Mar 16, 2022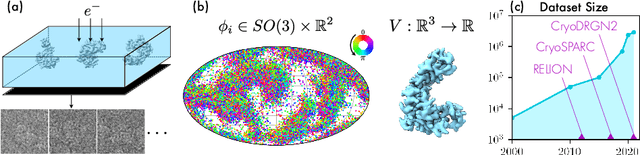
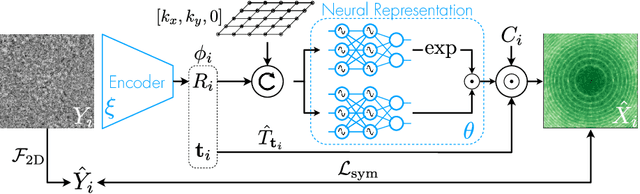
Cryo-electron microscopy (cryo-EM) has become a tool of fundamental importance in structural biology, helping us understand the basic building blocks of life. The algorithmic challenge of cryo-EM is to jointly estimate the unknown 3D poses and the 3D electron scattering potential of a biomolecule from millions of extremely noisy 2D images. Existing reconstruction algorithms, however, cannot easily keep pace with the rapidly growing size of cryo-EM datasets due to their high computational and memory cost. We introduce cryoAI, an ab initio reconstruction algorithm for homogeneous conformations that uses direct gradient-based optimization of particle poses and the electron scattering potential from single-particle cryo-EM data. CryoAI combines a learned encoder that predicts the poses of each particle image with a physics-based decoder to aggregate each particle image into an implicit representation of the scattering potential volume. This volume is stored in the Fourier domain for computational efficiency and leverages a modern coordinate network architecture for memory efficiency. Combined with a symmetrized loss function, this framework achieves results of a quality on par with state-of-the-art cryo-EM solvers for both simulated and experimental data, one order of magnitude faster for large datasets and with significantly lower memory requirements than existing methods.