 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAmortized Inference for Heterogeneous Reconstruction in Cryo-EM
Oct 13, 2022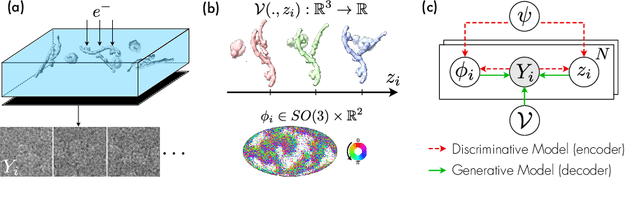
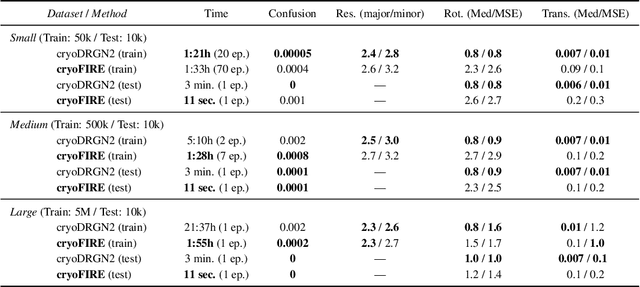
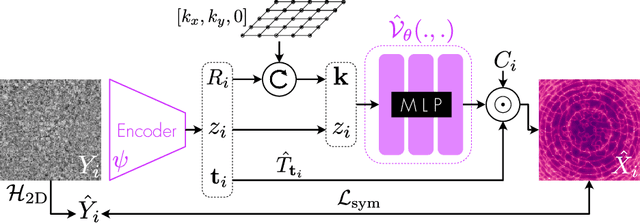

Cryo-electron microscopy (cryo-EM) is an imaging modality that provides unique insights into the dynamics of proteins and other building blocks of life. The algorithmic challenge of jointly estimating the poses, 3D structure, and conformational heterogeneity of a biomolecule from millions of noisy and randomly oriented 2D projections in a computationally efficient manner, however, remains unsolved. Our method, cryoFIRE, performs ab initio heterogeneous reconstruction with unknown poses in an amortized framework, thereby avoiding the computationally expensive step of pose search while enabling the analysis of conformational heterogeneity. Poses and conformation are jointly estimated by an encoder while a physics-based decoder aggregates the images into an implicit neural representation of the conformational space. We show that our method can provide one order of magnitude speedup on datasets containing millions of images without any loss of accuracy. We validate that the joint estimation of poses and conformations can be amortized over the size of the dataset. For the first time, we prove that an amortized method can extract interpretable dynamic information from experimental datasets.
Deep Generative Modeling for Volume Reconstruction in Cryo-Electron Microscopy
Jan 11, 2022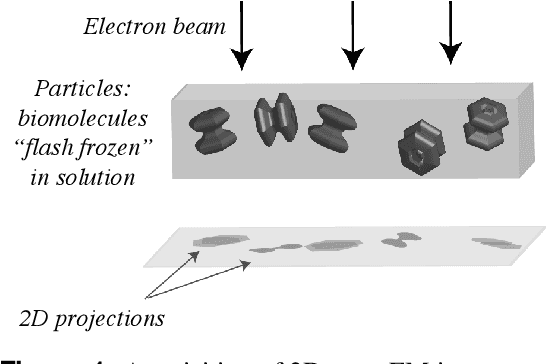

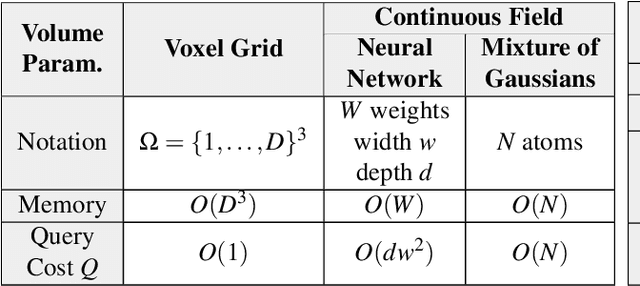

Recent breakthroughs in high resolution imaging of biomolecules in solution with cryo-electron microscopy (cryo-EM) have unlocked new doors for the reconstruction of molecular volumes, thereby promising further advances in biology, chemistry, and pharmacological research amongst others. Despite significant headway, the immense challenges in cryo-EM data analysis remain legion and intricately inter-disciplinary in nature, requiring insights from physicists, structural biologists, computer scientists, statisticians, and applied mathematicians. Meanwhile, recent next-generation volume reconstruction algorithms that combine generative modeling with end-to-end unsupervised deep learning techniques have shown promising results on simulated data, but still face considerable hurdles when applied to experimental cryo-EM images. In light of the proliferation of such methods and given the interdisciplinary nature of the task, we propose here a critical review of recent advances in the field of deep generative modeling for high resolution cryo-EM volume reconstruction. The present review aims to (i) compare and contrast these new methods, while (ii) presenting them from a perspective and using terminology familiar to scientists in each of the five aforementioned fields with no specific background in cryo-EM. The review begins with an introduction to the mathematical and computational challenges of deep generative models for cryo-EM volume reconstruction, along with an overview of the baseline methodology shared across this class of algorithms. Having established the common thread weaving through these different models, we provide a practical comparison of these state-of-the-art algorithms, highlighting their relative strengths and weaknesses, along with the assumptions that they rely on. This allows us to identify bottlenecks in current methods and avenues for future research.
End-to-End Simultaneous Learning of Single-particle Orientation and 3D Map Reconstruction from Cryo-electron Microscopy Data
Jul 07, 2021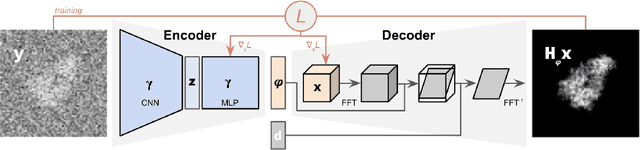
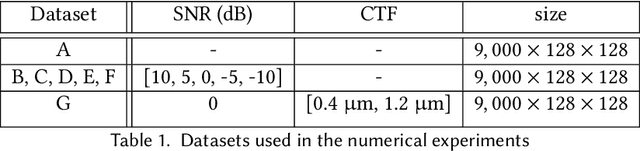
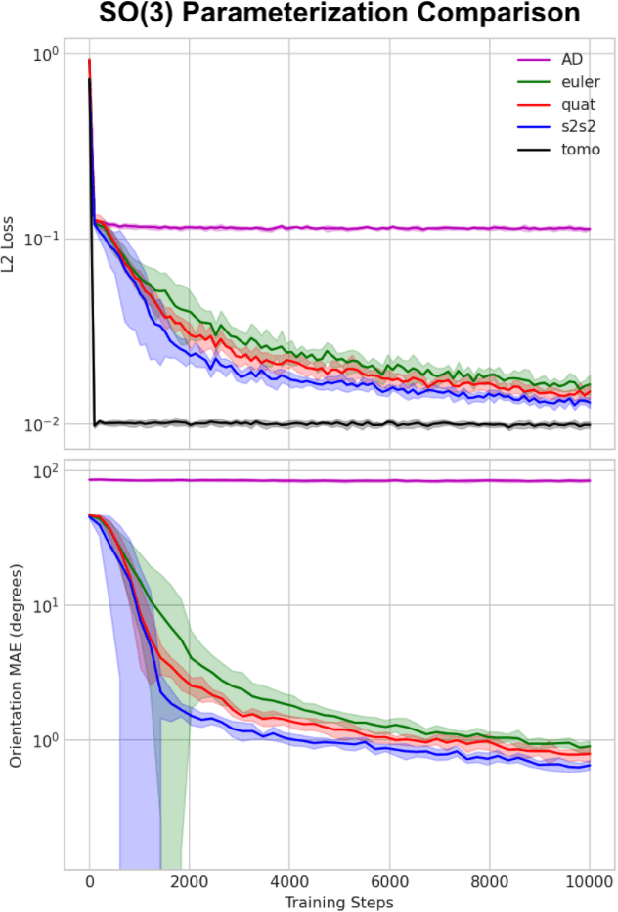
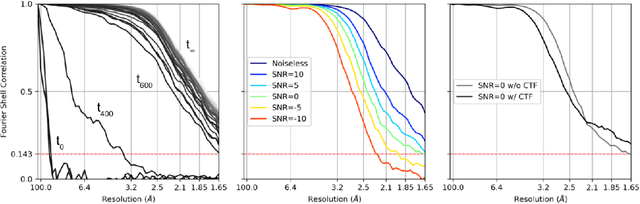
Cryogenic electron microscopy (cryo-EM) provides images from different copies of the same biomolecule in arbitrary orientations. Here, we present an end-to-end unsupervised approach that learns individual particle orientations from cryo-EM data while reconstructing the average 3D map of the biomolecule, starting from a random initialization. The approach relies on an auto-encoder architecture where the latent space is explicitly interpreted as orientations used by the decoder to form an image according to the linear projection model. We evaluate our method on simulated data and show that it is able to reconstruct 3D particle maps from noisy- and CTF-corrupted 2D projection images of unknown particle orientations.