 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultiscale guidance of AlphaFold3 with heterogeneous cryo-EM data
Jun 04, 2025Protein structure prediction models are now capable of generating accurate 3D structural hypotheses from sequence alone. However, they routinely fail to capture the conformational diversity of dynamic biomolecular complexes, often requiring heuristic MSA subsampling approaches for generating alternative states. In parallel, cryo-electron microscopy (cryo-EM) has emerged as a powerful tool for imaging near-native structural heterogeneity, but is challenged by arduous pipelines to go from raw experimental data to atomic models. Here, we bridge the gap between these modalities, combining cryo-EM density maps with the rich sequence and biophysical priors learned by protein structure prediction models. Our method, CryoBoltz, guides the sampling trajectory of a pretrained protein structure prediction model using both global and local structural constraints derived from density maps, driving predictions towards conformational states consistent with the experimental data. We demonstrate that this flexible yet powerful inference-time approach allows us to build atomic models into heterogeneous cryo-EM maps across a variety of dynamic biomolecular systems including transporters and antibodies.
Mixture of neural fields for heterogeneous reconstruction in cryo-EM
Dec 12, 2024



Cryo-electron microscopy (cryo-EM) is an experimental technique for protein structure determination that images an ensemble of macromolecules in near-physiological contexts. While recent advances enable the reconstruction of dynamic conformations of a single biomolecular complex, current methods do not adequately model samples with mixed conformational and compositional heterogeneity. In particular, datasets containing mixtures of multiple proteins require the joint inference of structure, pose, compositional class, and conformational states for 3D reconstruction. Here, we present Hydra, an approach that models both conformational and compositional heterogeneity fully ab initio by parameterizing structures as arising from one of K neural fields. We employ a new likelihood-based loss function and demonstrate the effectiveness of our approach on synthetic datasets composed of mixtures of proteins with large degrees of conformational variability. We additionally demonstrate Hydra on an experimental dataset of a cellular lysate containing a mixture of different protein complexes. Hydra expands the expressivity of heterogeneous reconstruction methods and thus broadens the scope of cryo-EM to increasingly complex samples.
CryoBench: Diverse and challenging datasets for the heterogeneity problem in cryo-EM
Aug 10, 2024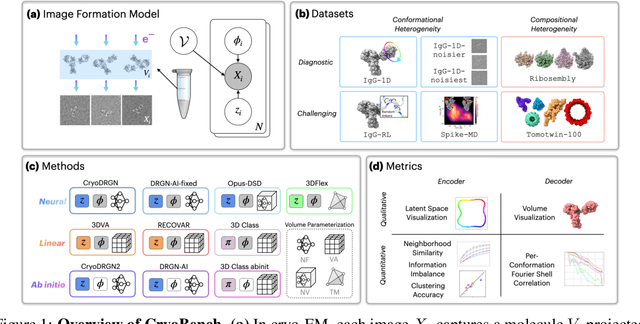
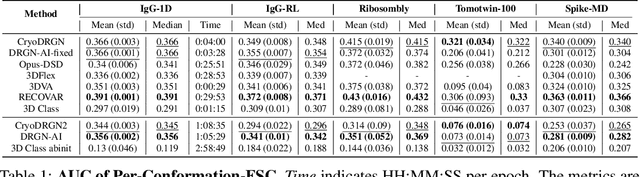
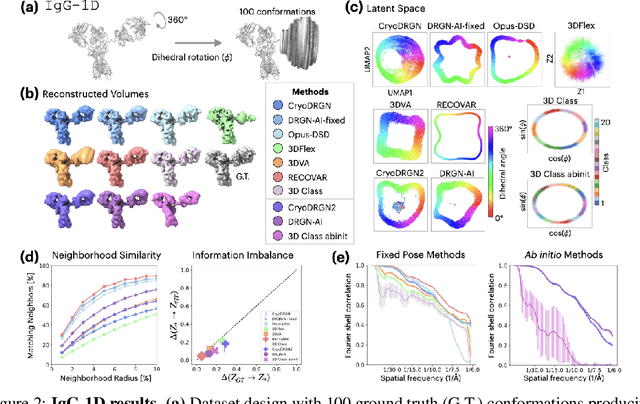

Cryo-electron microscopy (cryo-EM) is a powerful technique for determining high-resolution 3D biomolecular structures from imaging data. As this technique can capture dynamic biomolecular complexes, 3D reconstruction methods are increasingly being developed to resolve this intrinsic structural heterogeneity. However, the absence of standardized benchmarks with ground truth structures and validation metrics limits the advancement of the field. Here, we propose CryoBench, a suite of datasets, metrics, and performance benchmarks for heterogeneous reconstruction in cryo-EM. We propose five datasets representing different sources of heterogeneity and degrees of difficulty. These include conformational heterogeneity generated from simple motions and random configurations of antibody complexes and from tens of thousands of structures sampled from a molecular dynamics simulation. We also design datasets containing compositional heterogeneity from mixtures of ribosome assembly states and 100 common complexes present in cells. We then perform a comprehensive analysis of state-of-the-art heterogeneous reconstruction tools including neural and non-neural methods and their sensitivity to noise, and propose new metrics for quantitative comparison of methods. We hope that this benchmark will be a foundational resource for analyzing existing methods and new algorithmic development in both the cryo-EM and machine learning communities.