 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRobust Inference-Time Steering of Protein Diffusion Models via Embedding Optimization
Feb 05, 2026In many biophysical inverse problems, the goal is to generate biomolecular conformations that are both physically plausible and consistent with experimental measurements. As recent sequence-to-structure diffusion models provide powerful data-driven priors, posterior sampling has emerged as a popular framework by guiding atomic coordinates to target conformations using experimental likelihoods. However, when the target lies in a low-density region of the prior, posterior sampling requires aggressive and brittle weighting of the likelihood guidance. Motivated by this limitation, we propose EmbedOpt, an alternative inference-time approach for steering diffusion models to optimize experimental likelihoods in the conditional embedding space. As this space encodes rich sequence and coevolutionary signals, optimizing over it effectively shifts the diffusion prior to align with experimental constraints. We validate EmbedOpt on two benchmarks simulating cryo-electron microscopy map fitting and experimental distance constraints. We show that EmbedOpt outperforms the coordinate-based posterior sampling method in map fitting tasks, matches performance on distance constraint tasks, and exhibits superior engineering robustness across hyperparameters spanning two orders of magnitude. Moreover, its smooth optimization behavior enables a significant reduction in the number of diffusion steps required for inference, leading to better efficiency.
Cryo-em images are intrinsically low dimensional
Apr 15, 2025Simulation-based inference provides a powerful framework for cryo-electron microscopy, employing neural networks in methods like CryoSBI to infer biomolecular conformations via learned latent representations. This latent space represents a rich opportunity, encoding valuable information about the physical system and the inference process. Harnessing this potential hinges on understanding the underlying geometric structure of these representations. We investigate this structure by applying manifold learning techniques to CryoSBI representations of hemagglutinin (simulated and experimental). We reveal that these high-dimensional data inherently populate low-dimensional, smooth manifolds, with simulated data effectively covering the experimental counterpart. By characterizing the manifold's geometry using Diffusion Maps and identifying its principal axes of variation via coordinate interpretation methods, we establish a direct link between the latent structure and key physical parameters. Discovering this intrinsic low-dimensionality and interpretable geometric organization not only validates the CryoSBI approach but enables us to learn more from the data structure and provides opportunities for improving future inference strategies by exploiting this revealed manifold geometry.
CryoBench: Diverse and challenging datasets for the heterogeneity problem in cryo-EM
Aug 10, 2024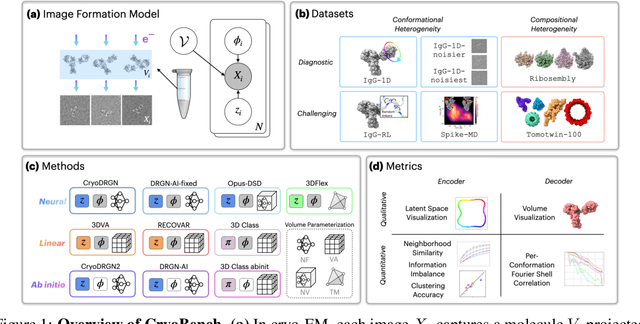
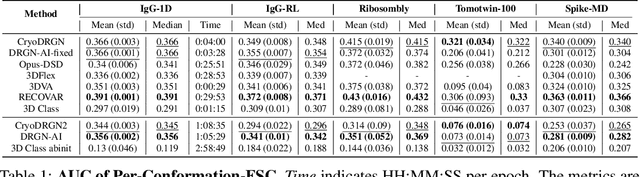
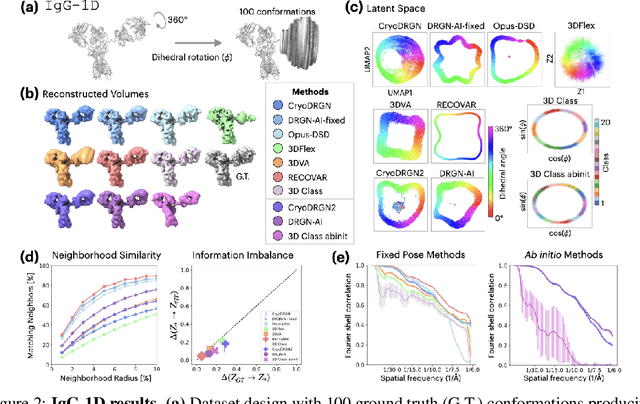

Cryo-electron microscopy (cryo-EM) is a powerful technique for determining high-resolution 3D biomolecular structures from imaging data. As this technique can capture dynamic biomolecular complexes, 3D reconstruction methods are increasingly being developed to resolve this intrinsic structural heterogeneity. However, the absence of standardized benchmarks with ground truth structures and validation metrics limits the advancement of the field. Here, we propose CryoBench, a suite of datasets, metrics, and performance benchmarks for heterogeneous reconstruction in cryo-EM. We propose five datasets representing different sources of heterogeneity and degrees of difficulty. These include conformational heterogeneity generated from simple motions and random configurations of antibody complexes and from tens of thousands of structures sampled from a molecular dynamics simulation. We also design datasets containing compositional heterogeneity from mixtures of ribosome assembly states and 100 common complexes present in cells. We then perform a comprehensive analysis of state-of-the-art heterogeneous reconstruction tools including neural and non-neural methods and their sensitivity to noise, and propose new metrics for quantitative comparison of methods. We hope that this benchmark will be a foundational resource for analyzing existing methods and new algorithmic development in both the cryo-EM and machine learning communities.
Active learning of Boltzmann samplers and potential energies with quantum mechanical accuracy
Jan 29, 2024



Extracting consistent statistics between relevant free-energy minima of a molecular system is essential for physics, chemistry and biology. Molecular dynamics (MD) simulations can aid in this task but are computationally expensive, especially for systems that require quantum accuracy. To overcome this challenge, we develop an approach combining enhanced sampling with deep generative models and active learning of a machine learning potential (MLP). We introduce an adaptive Markov chain Monte Carlo framework that enables the training of one Normalizing Flow (NF) and one MLP per state. We simulate several Markov chains in parallel until they reach convergence, sampling the Boltzmann distribution with an efficient use of energy evaluations. At each iteration, we compute the energy of a subset of the NF-generated configurations using Density Functional Theory (DFT), we predict the remaining configuration's energy with the MLP and actively train the MLP using the DFT-computed energies. Leveraging the trained NF and MLP models, we can compute thermodynamic observables such as free-energy differences or optical spectra. We apply this method to study the isomerization of an ultrasmall silver nanocluster, belonging to a set of systems with diverse applications in the fields of medicine and catalysis.