 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTICON: A Slide-Level Tile Contextualizer for Histopathology Representation Learning
Dec 24, 2025The interpretation of small tiles in large whole slide images (WSI) often needs a larger image context. We introduce TICON, a transformer-based tile representation contextualizer that produces rich, contextualized embeddings for ''any'' application in computational pathology. Standard tile encoder-based pipelines, which extract embeddings of tiles stripped from their context, fail to model the rich slide-level information essential for both local and global tasks. Furthermore, different tile-encoders excel at different downstream tasks. Therefore, a unified model is needed to contextualize embeddings derived from ''any'' tile-level foundation model. TICON addresses this need with a single, shared encoder, pretrained using a masked modeling objective to simultaneously unify and contextualize representations from diverse tile-level pathology foundation models. Our experiments demonstrate that TICON-contextualized embeddings significantly improve performance across many different tasks, establishing new state-of-the-art results on tile-level benchmarks (i.e., HEST-Bench, THUNDER, CATCH) and slide-level benchmarks (i.e., Patho-Bench). Finally, we pretrain an aggregator on TICON to form a slide-level foundation model, using only 11K WSIs, outperforming SoTA slide-level foundation models pretrained with up to 350K WSIs.
PixCell: A generative foundation model for digital histopathology images
Jun 05, 2025The digitization of histology slides has revolutionized pathology, providing massive datasets for cancer diagnosis and research. Contrastive self-supervised and vision-language models have been shown to effectively mine large pathology datasets to learn discriminative representations. On the other hand, generative models, capable of synthesizing realistic and diverse images, present a compelling solution to address unique problems in pathology that involve synthesizing images; overcoming annotated data scarcity, enabling privacy-preserving data sharing, and performing inherently generative tasks, such as virtual staining. We introduce PixCell, the first diffusion-based generative foundation model for histopathology. We train PixCell on PanCan-30M, a vast, diverse dataset derived from 69,184 H\&E-stained whole slide images covering various cancer types. We employ a progressive training strategy and a self-supervision-based conditioning that allows us to scale up training without any annotated data. PixCell generates diverse and high-quality images across multiple cancer types, which we find can be used in place of real data to train a self-supervised discriminative model. Synthetic images shared between institutions are subject to fewer regulatory barriers than would be the case with real clinical images. Furthermore, we showcase the ability to precisely control image generation using a small set of annotated images, which can be used for both data augmentation and educational purposes. Testing on a cell segmentation task, a mask-guided PixCell enables targeted data augmentation, improving downstream performance. Finally, we demonstrate PixCell's ability to use H\&E structural staining to infer results from molecular marker studies; we use this capability to infer IHC staining from H\&E images. Our trained models are publicly released to accelerate research in computational pathology.
GECKO: Gigapixel Vision-Concept Contrastive Pretraining in Histopathology
Apr 01, 2025Pretraining a Multiple Instance Learning (MIL) aggregator enables the derivation of Whole Slide Image (WSI)-level embeddings from patch-level representations without supervision. While recent multimodal MIL pretraining approaches leveraging auxiliary modalities have demonstrated performance gains over unimodal WSI pretraining, the acquisition of these additional modalities necessitates extensive clinical profiling. This requirement increases costs and limits scalability in existing WSI datasets lacking such paired modalities. To address this, we propose Gigapixel Vision-Concept Knowledge Contrastive pretraining (GECKO), which aligns WSIs with a Concept Prior derived from the available WSIs. First, we derive an inherently interpretable concept prior by computing the similarity between each WSI patch and textual descriptions of predefined pathology concepts. GECKO then employs a dual-branch MIL network: one branch aggregates patch embeddings into a WSI-level deep embedding, while the other aggregates the concept prior into a corresponding WSI-level concept embedding. Both aggregated embeddings are aligned using a contrastive objective, thereby pretraining the entire dual-branch MIL model. Moreover, when auxiliary modalities such as transcriptomics data are available, GECKO seamlessly integrates them. Across five diverse tasks, GECKO consistently outperforms prior unimodal and multimodal pretraining approaches while also delivering clinically meaningful interpretability that bridges the gap between computational models and pathology expertise. Code is made available at https://github.com/bmi-imaginelab/GECKO
Gen-SIS: Generative Self-augmentation Improves Self-supervised Learning
Dec 02, 2024



Self-supervised learning (SSL) methods have emerged as strong visual representation learners by training an image encoder to maximize similarity between features of different views of the same image. To perform this view-invariance task, current SSL algorithms rely on hand-crafted augmentations such as random cropping and color jittering to create multiple views of an image. Recently, generative diffusion models have been shown to improve SSL by providing a wider range of data augmentations. However, these diffusion models require pre-training on large-scale image-text datasets, which might not be available for many specialized domains like histopathology. In this work, we introduce Gen-SIS, a diffusion-based augmentation technique trained exclusively on unlabeled image data, eliminating any reliance on external sources of supervision such as text captions. We first train an initial SSL encoder on a dataset using only hand-crafted augmentations. We then train a diffusion model conditioned on embeddings from that SSL encoder. Following training, given an embedding of the source image, this diffusion model can synthesize its diverse views. We show that these `self-augmentations', i.e. generative augmentations based on the vanilla SSL encoder embeddings, facilitate the training of a stronger SSL encoder. Furthermore, based on the ability to interpolate between images in the encoder latent space, we introduce the novel pretext task of disentangling the two source images of an interpolated synthetic image. We validate Gen-SIS's effectiveness by demonstrating performance improvements across various downstream tasks in both natural images, which are generally object-centric, as well as digital histopathology images, which are typically context-based.
RankByGene: Gene-Guided Histopathology Representation Learning Through Cross-Modal Ranking Consistency
Nov 22, 2024Spatial transcriptomics (ST) provides essential spatial context by mapping gene expression within tissue, enabling detailed study of cellular heterogeneity and tissue organization. However, aligning ST data with histology images poses challenges due to inherent spatial distortions and modality-specific variations. Existing methods largely rely on direct alignment, which often fails to capture complex cross-modal relationships. To address these limitations, we propose a novel framework that aligns gene and image features using a ranking-based alignment loss, preserving relative similarity across modalities and enabling robust multi-scale alignment. To further enhance the alignment's stability, we employ self-supervised knowledge distillation with a teacher-student network architecture, effectively mitigating disruptions from high dimensionality, sparsity, and noise in gene expression data. Extensive experiments on gene expression prediction and survival analysis demonstrate our framework's effectiveness, showing improved alignment and predictive performance over existing methods and establishing a robust tool for gene-guided image representation learning in digital pathology.
Histo-Diffusion: A Diffusion Super-Resolution Method for Digital Pathology with Comprehensive Quality Assessment
Aug 27, 2024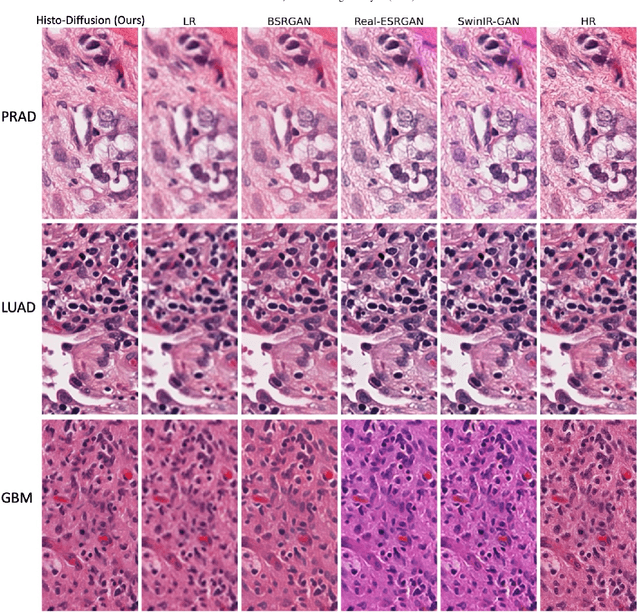
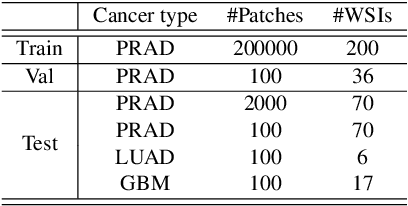
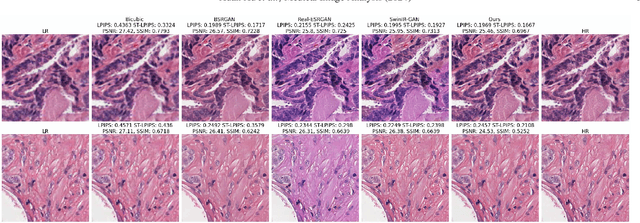
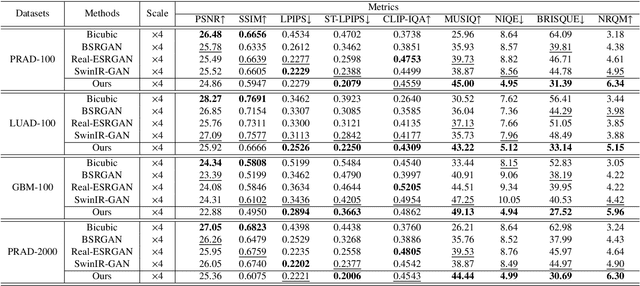
Digital pathology has advanced significantly over the last decade, with Whole Slide Images (WSIs) encompassing vast amounts of data essential for accurate disease diagnosis. High-resolution WSIs are essential for precise diagnosis but technical limitations in scanning equipment and variablity in slide preparation can hinder obtaining these images. Super-resolution techniques can enhance low-resolution images; while Generative Adversarial Networks (GANs) have been effective in natural image super-resolution tasks, they often struggle with histopathology due to overfitting and mode collapse. Traditional evaluation metrics fall short in assessing the complex characteristics of histopathology images, necessitating robust histology-specific evaluation methods. We introduce Histo-Diffusion, a novel diffusion-based method specially designed for generating and evaluating super-resolution images in digital pathology. It includes a restoration module for histopathology prior and a controllable diffusion module for generating high-quality images. We have curated two histopathology datasets and proposed a comprehensive evaluation strategy which incorporates both full-reference and no-reference metrics to thoroughly assess the quality of digital pathology images. Comparative analyses on multiple datasets with state-of-the-art methods reveal that Histo-Diffusion outperforms GANs. Our method offers a versatile solution for histopathology image super-resolution, capable of handling multi-resolution generation from varied input sizes, providing valuable support in diagnostic processes.
SI-MIL: Taming Deep MIL for Self-Interpretability in Gigapixel Histopathology
Dec 22, 2023



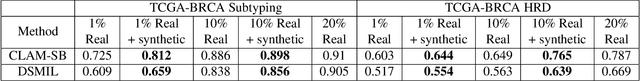
Introducing interpretability and reasoning into Multiple Instance Learning (MIL) methods for Whole Slide Image (WSI) analysis is challenging, given the complexity of gigapixel slides. Traditionally, MIL interpretability is limited to identifying salient regions deemed pertinent for downstream tasks, offering little insight to the end-user (pathologist) regarding the rationale behind these selections. To address this, we propose Self-Interpretable MIL (SI-MIL), a method intrinsically designed for interpretability from the very outset. SI-MIL employs a deep MIL framework to guide an interpretable branch grounded on handcrafted pathological features, facilitating linear predictions. Beyond identifying salient regions, SI-MIL uniquely provides feature-level interpretations rooted in pathological insights for WSIs. Notably, SI-MIL, with its linear prediction constraints, challenges the prevalent myth of an inevitable trade-off between model interpretability and performance, demonstrating competitive results compared to state-of-the-art methods on WSI-level prediction tasks across three cancer types. In addition, we thoroughly benchmark the local- and global-interpretability of SI-MIL in terms of statistical analysis, a domain expert study, and desiderata of interpretability, namely, user-friendliness and faithfulness.
Learned representation-guided diffusion models for large-image generation
Dec 12, 2023
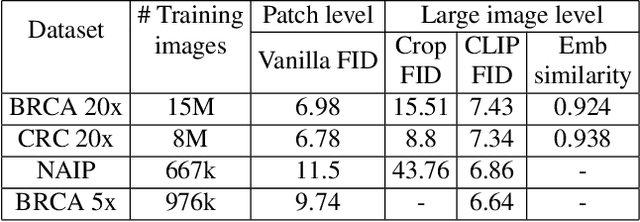

To synthesize high-fidelity samples, diffusion models typically require auxiliary data to guide the generation process. However, it is impractical to procure the painstaking patch-level annotation effort required in specialized domains like histopathology and satellite imagery; it is often performed by domain experts and involves hundreds of millions of patches. Modern-day self-supervised learning (SSL) representations encode rich semantic and visual information. In this paper, we posit that such representations are expressive enough to act as proxies to fine-grained human labels. We introduce a novel approach that trains diffusion models conditioned on embeddings from SSL. Our diffusion models successfully project these features back to high-quality histopathology and remote sensing images. In addition, we construct larger images by assembling spatially consistent patches inferred from SSL embeddings, preserving long-range dependencies. Augmenting real data by generating variations of real images improves downstream classifier accuracy for patch-level and larger, image-scale classification tasks. Our models are effective even on datasets not encountered during training, demonstrating their robustness and generalizability. Generating images from learned embeddings is agnostic to the source of the embeddings. The SSL embeddings used to generate a large image can either be extracted from a reference image, or sampled from an auxiliary model conditioned on any related modality (e.g. class labels, text, genomic data). As proof of concept, we introduce the text-to-large image synthesis paradigm where we successfully synthesize large pathology and satellite images out of text descriptions.
Attention De-sparsification Matters: Inducing Diversity in Digital Pathology Representation Learning
Sep 12, 2023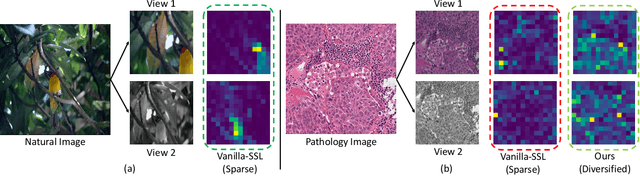
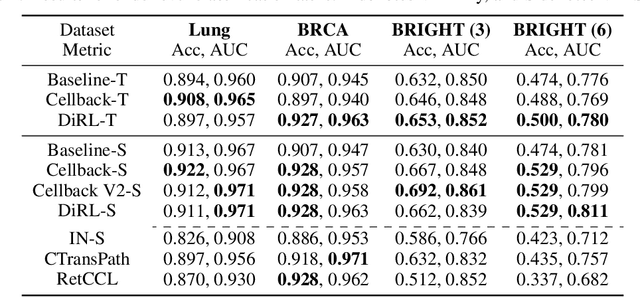
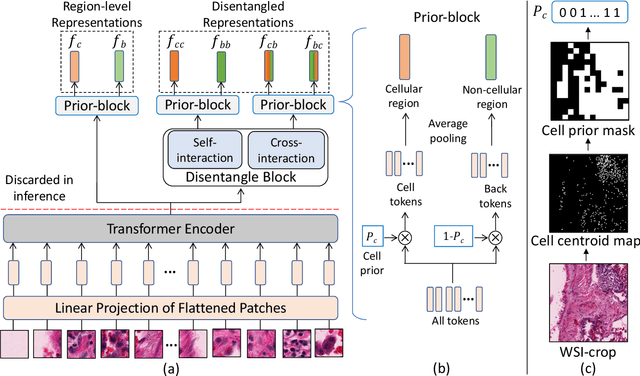
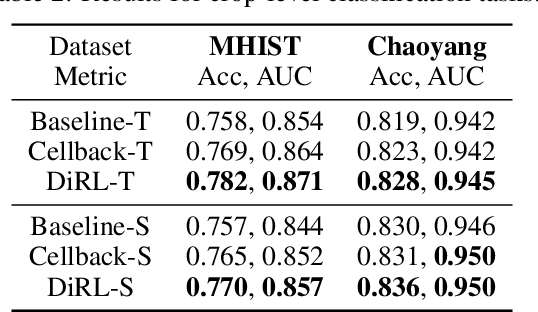
We propose DiRL, a Diversity-inducing Representation Learning technique for histopathology imaging. Self-supervised learning techniques, such as contrastive and non-contrastive approaches, have been shown to learn rich and effective representations of digitized tissue samples with limited pathologist supervision. Our analysis of vanilla SSL-pretrained models' attention distribution reveals an insightful observation: sparsity in attention, i.e, models tends to localize most of their attention to some prominent patterns in the image. Although attention sparsity can be beneficial in natural images due to these prominent patterns being the object of interest itself, this can be sub-optimal in digital pathology; this is because, unlike natural images, digital pathology scans are not object-centric, but rather a complex phenotype of various spatially intermixed biological components. Inadequate diversification of attention in these complex images could result in crucial information loss. To address this, we leverage cell segmentation to densely extract multiple histopathology-specific representations, and then propose a prior-guided dense pretext task for SSL, designed to match the multiple corresponding representations between the views. Through this, the model learns to attend to various components more closely and evenly, thus inducing adequate diversification in attention for capturing context rich representations. Through quantitative and qualitative analysis on multiple tasks across cancer types, we demonstrate the efficacy of our method and observe that the attention is more globally distributed.
SAM-Path: A Segment Anything Model for Semantic Segmentation in Digital Pathology
Jul 12, 2023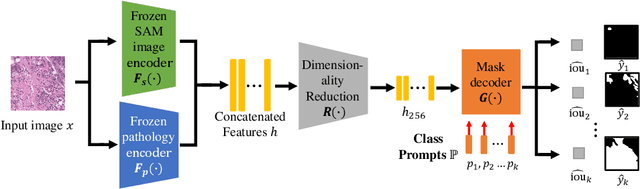
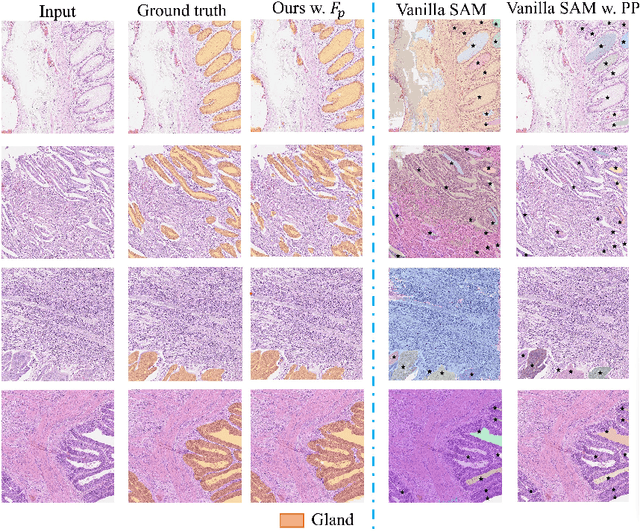
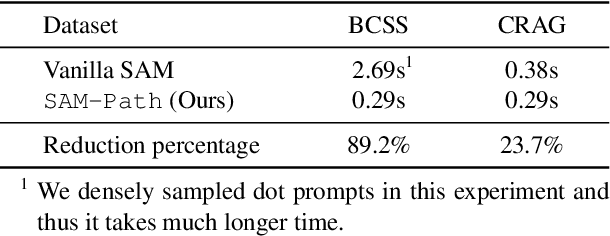
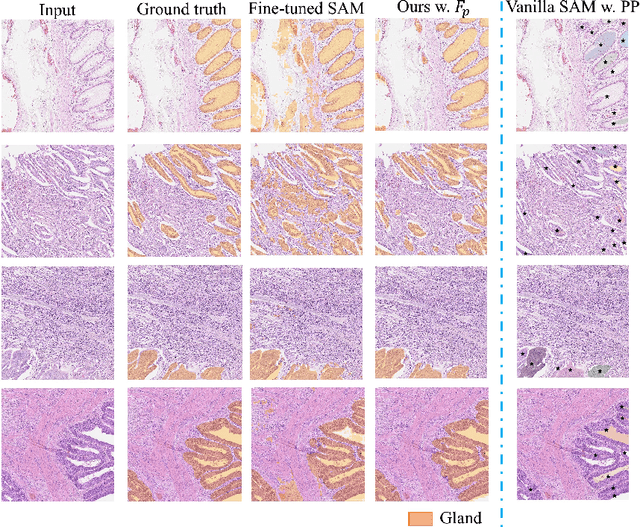
Semantic segmentations of pathological entities have crucial clinical value in computational pathology workflows. Foundation models, such as the Segment Anything Model (SAM), have been recently proposed for universal use in segmentation tasks. SAM shows remarkable promise in instance segmentation on natural images. However, the applicability of SAM to computational pathology tasks is limited due to the following factors: (1) lack of comprehensive pathology datasets used in SAM training and (2) the design of SAM is not inherently optimized for semantic segmentation tasks. In this work, we adapt SAM for semantic segmentation by introducing trainable class prompts, followed by further enhancements through the incorporation of a pathology encoder, specifically a pathology foundation model. Our framework, SAM-Path enhances SAM's ability to conduct semantic segmentation in digital pathology without human input prompts. Through experiments on two public pathology datasets, the BCSS and the CRAG datasets, we demonstrate that the fine-tuning with trainable class prompts outperforms vanilla SAM with manual prompts and post-processing by 27.52% in Dice score and 71.63% in IOU. On these two datasets, the proposed additional pathology foundation model further achieves a relative improvement of 5.07% to 5.12% in Dice score and 4.50% to 8.48% in IOU.