 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTopo-R1: Detecting Topological Anomalies via Vision-Language Models
Mar 13, 2026Topological correctness is crucial for tubular structures such as blood vessels, nerve fibers, and road networks. Existing topology-preserving methods rely on domain-specific ground truth, which is costly and rarely transfers across domains. When deployed to a new domain without annotations, a key question arises: how can we detect topological anomalies without ground-truth supervision? We reframe this as topological anomaly detection, a structured visual reasoning task requiring a model to locate and classify topological errors in predicted segmentation masks. Vision-Language Models (VLMs) are natural candidates; however, we find that state-of-the-art VLMs perform nearly at random, lacking the fine-grained, topology-aware perception needed to identify sparse connectivity errors in dense structures. To bridge this gap, we develop an automated data-curation pipeline that synthesizes diverse topological anomalies with verifiable annotations across progressively difficult levels, thereby constructing the first large-scale, multi-domain benchmark for this task. We then introduce Topo-R1, a framework that endows VLMs with topology-aware perception via two-stage training: supervised fine-tuning followed by reinforcement learning with Group Relative Policy Optimization (GRPO). Central to our approach is a topology-aware composite reward that integrates type-aware Hungarian matching for structured error classification, spatial localization scoring, and a centerline Dice (clDice) reward that directly penalizes connectivity disruptions, thereby jointly incentivizing semantic precision and structural fidelity. Extensive experiments demonstrate that Topo-R1 establishes a new paradigm for annotation-free topological quality assessment, consistently outperforming general-purpose VLMs and supervised baselines across all evaluation protocols.
Act Like a Pathologist: Tissue-Aware Whole Slide Image Reasoning
Feb 28, 2026Computational pathology has advanced rapidly in recent years, driven by domain-specific image encoders and growing interest in using vision-language models to answer natural-language questions about diseases. Yet, the core problem behind pathology question-answering remains unsolved, considering that a gigapixel slide contains far more information than necessary for a given question. Pathologists naturally navigate tissue and morphology complexity by scanning broadly, and zooming in selectively according to the clinical questions. Current models, in contrast, rely on uniform patch sampling or broad attention maps, often attending equally to irrelevant regions while overlooking key visual evidence. In this work, we try to bring models closer to how humans actually examine slides. We propose a question-guided, tissue-aware, and coarse-to-fine retrieval framework, HistoSelect, that consists of two key components: a group sampler that identifies question-relevant tissue regions, followed by a patch selector that retrieves the most informative patches within those regions. By selecting only the most informative patches, our method becomes significantly more efficient: reducing visual token usage by 70% on average, while improving accuracy across three pathology QA tasks. Evaluated on 356,000 question-answer pairs, our approach outperforms existing methods and produces answers grounded in interpretable, pathologist-consistent regions. Our results suggest that bringing human-like search and attention patterns into WSI reasoning is a promising direction for building practical and reliable pathology VLMs.
MATCH: Multi-faceted Adaptive Topo-Consistency for Semi-Supervised Histopathology Segmentation
Oct 02, 2025



In semi-supervised segmentation, capturing meaningful semantic structures from unlabeled data is essential. This is particularly challenging in histopathology image analysis, where objects are densely distributed. To address this issue, we propose a semi-supervised segmentation framework designed to robustly identify and preserve relevant topological features. Our method leverages multiple perturbed predictions obtained through stochastic dropouts and temporal training snapshots, enforcing topological consistency across these varied outputs. This consistency mechanism helps distinguish biologically meaningful structures from transient and noisy artifacts. A key challenge in this process is to accurately match the corresponding topological features across the predictions in the absence of ground truth. To overcome this, we introduce a novel matching strategy that integrates spatial overlap with global structural alignment, minimizing discrepancies among predictions. Extensive experiments demonstrate that our approach effectively reduces topological errors, resulting in more robust and accurate segmentations essential for reliable downstream analysis. Code is available at \href{https://github.com/Melon-Xu/MATCH}{https://github.com/Melon-Xu/MATCH}.
TopoCellGen: Generating Histopathology Cell Topology with a Diffusion Model
Dec 08, 2024Accurately modeling multi-class cell topology is crucial in digital pathology, as it provides critical insights into tissue structure and pathology. The synthetic generation of cell topology enables realistic simulations of complex tissue environments, enhances downstream tasks by augmenting training data, aligns more closely with pathologists' domain knowledge, and offers new opportunities for controlling and generalizing the tumor microenvironment. In this paper, we propose a novel approach that integrates topological constraints into a diffusion model to improve the generation of realistic, contextually accurate cell topologies. Our method refines the simulation of cell distributions and interactions, increasing the precision and interpretability of results in downstream tasks such as cell detection and classification. To assess the topological fidelity of generated layouts, we introduce a new metric, Topological Frechet Distance (TopoFD), which overcomes the limitations of traditional metrics like FID in evaluating topological structure. Experimental results demonstrate the effectiveness of our approach in generating multi-class cell layouts that capture intricate topological relationships.
RankByGene: Gene-Guided Histopathology Representation Learning Through Cross-Modal Ranking Consistency
Nov 22, 2024Spatial transcriptomics (ST) provides essential spatial context by mapping gene expression within tissue, enabling detailed study of cellular heterogeneity and tissue organization. However, aligning ST data with histology images poses challenges due to inherent spatial distortions and modality-specific variations. Existing methods largely rely on direct alignment, which often fails to capture complex cross-modal relationships. To address these limitations, we propose a novel framework that aligns gene and image features using a ranking-based alignment loss, preserving relative similarity across modalities and enabling robust multi-scale alignment. To further enhance the alignment's stability, we employ self-supervised knowledge distillation with a teacher-student network architecture, effectively mitigating disruptions from high dimensionality, sparsity, and noise in gene expression data. Extensive experiments on gene expression prediction and survival analysis demonstrate our framework's effectiveness, showing improved alignment and predictive performance over existing methods and establishing a robust tool for gene-guided image representation learning in digital pathology.
Hard Negative Sample Mining for Whole Slide Image Classification
Oct 03, 2024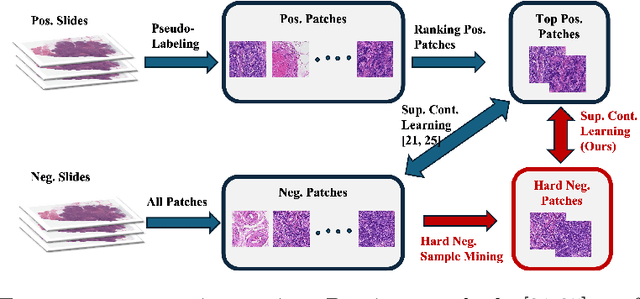
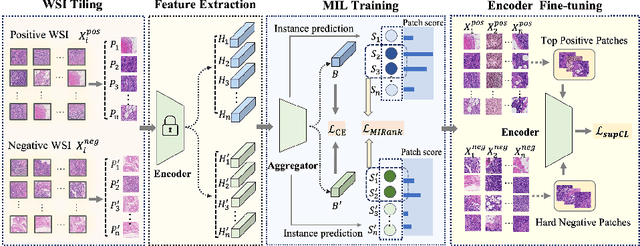
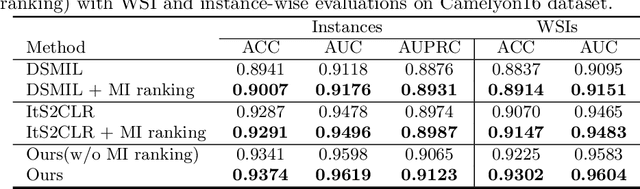
Weakly supervised whole slide image (WSI) classification is challenging due to the lack of patch-level labels and high computational costs. State-of-the-art methods use self-supervised patch-wise feature representations for multiple instance learning (MIL). Recently, methods have been proposed to fine-tune the feature representation on the downstream task using pseudo labeling, but mostly focusing on selecting high-quality positive patches. In this paper, we propose to mine hard negative samples during fine-tuning. This allows us to obtain better feature representations and reduce the training cost. Furthermore, we propose a novel patch-wise ranking loss in MIL to better exploit these hard negative samples. Experiments on two public datasets demonstrate the efficacy of these proposed ideas. Our codes are available at https://github.com/winston52/HNM-WSI
Semi-Supervised Contrastive VAE for Disentanglement of Digital Pathology Images
Oct 02, 2024Despite the strong prediction power of deep learning models, their interpretability remains an important concern. Disentanglement models increase interpretability by decomposing the latent space into interpretable subspaces. In this paper, we propose the first disentanglement method for pathology images. We focus on the task of detecting tumor-infiltrating lymphocytes (TIL). We propose different ideas including cascading disentanglement, novel architecture, and reconstruction branches. We achieve superior performance on complex pathology images, thus improving the interpretability and even generalization power of TIL detection deep learning models. Our codes are available at https://github.com/Shauqi/SS-cVAE.
Spatial Diffusion for Cell Layout Generation
Sep 04, 2024



Generative models, such as GANs and diffusion models, have been used to augment training sets and boost performances in different tasks. We focus on generative models for cell detection instead, i.e., locating and classifying cells in given pathology images. One important information that has been largely overlooked is the spatial patterns of the cells. In this paper, we propose a spatial-pattern-guided generative model for cell layout generation. Specifically, a novel diffusion model guided by spatial features and generates realistic cell layouts has been proposed. We explore different density models as spatial features for the diffusion model. In downstream tasks, we show that the generated cell layouts can be used to guide the generation of high-quality pathology images. Augmenting with these images can significantly boost the performance of SOTA cell detection methods. The code is available at https://github.com/superlc1995/Diffusion-cell.
TopoSemiSeg: Enforcing Topological Consistency for Semi-Supervised Segmentation of Histopathology Images
Dec 06, 2023In computational pathology, segmenting densely distributed objects like glands and nuclei is crucial for downstream analysis. To alleviate the burden of obtaining pixel-wise annotations, semi-supervised learning methods learn from large amounts of unlabeled data. Nevertheless, existing semi-supervised methods overlook the topological information hidden in the unlabeled images and are thus prone to topological errors, e.g., missing or incorrectly merged/separated glands or nuclei. To address this issue, we propose TopoSemiSeg, the first semi-supervised method that learns the topological representation from unlabeled data. In particular, we propose a topology-aware teacher-student approach in which the teacher and student networks learn shared topological representations. To achieve this, we introduce topological consistency loss, which contains signal consistency and noise removal losses to ensure the learned representation is robust and focuses on true topological signals. Extensive experiments on public pathology image datasets show the superiority of our method, especially on topology-wise evaluation metrics. Code is available at https://github.com/Melon-Xu/TopoSemiSeg.
Calibrating Uncertainty for Semi-Supervised Crowd Counting
Aug 19, 2023



Semi-supervised crowd counting is an important yet challenging task. A popular approach is to iteratively generate pseudo-labels for unlabeled data and add them to the training set. The key is to use uncertainty to select reliable pseudo-labels. In this paper, we propose a novel method to calibrate model uncertainty for crowd counting. Our method takes a supervised uncertainty estimation strategy to train the model through a surrogate function. This ensures the uncertainty is well controlled throughout the training. We propose a matching-based patch-wise surrogate function to better approximate uncertainty for crowd counting tasks. The proposed method pays a sufficient amount of attention to details, while maintaining a proper granularity. Altogether our method is able to generate reliable uncertainty estimation, high quality pseudolabels, and achieve state-of-the-art performance in semisupervised crowd counting.