 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDialysis Risk Prediction and Treatment Effect Estimation for AKI patients using Longitudinal Electronic Health Records
Apr 27, 2026Progression to dialysis or end-stage renal disease is a rare but clinically important outcome. Clinicians need evidence on how medication exposures influence downstream risk. We constructed a fixed-window EHR cohort (90-day observation, 730-day prediction; N=81401; dialysis/ESRD prevalence: 1.1%) and modeled sequences of diagnoses, procedures, and medications with kidney laboratory trends (creatinine, BUN, eGFR). A transformer-based causal multi-head model was trained to estimate drug- and ingredient-level average treatment effects (ATEs) using counterfactual exposure removal and insertion under a full medication history setup. On test set, predictive performance reached an AUC of 0.694 and PR-AUC of 0.094. At the selected decision threshold (0.883), the model achieved an F1 score of 0.201 with a Brier score of 0.018. Post-hoc causal analyses of lab changes (eGFR, creatinine, BUN) using IPTW, AIPW, naive, and covariate-adjusted OLS methods assessed clinical directionality. Results showed partial protective-direction support for ACE/ARB exposures and worsening-direction signals for loop diuretics.
DeepVRegulome: DNABERT-based deep-learning framework for predicting the functional impact of short genomic variants on the human regulome
Nov 12, 2025
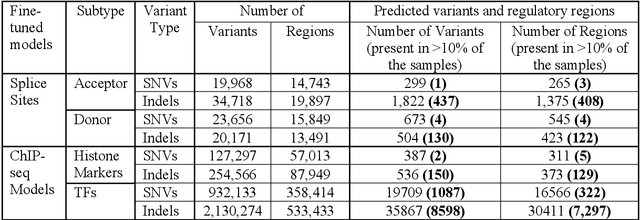


Whole-genome sequencing (WGS) has revealed numerous non-coding short variants whose functional impacts remain poorly understood. Despite recent advances in deep-learning genomic approaches, accurately predicting and prioritizing clinically relevant mutations in gene regulatory regions remains a major challenge. Here we introduce Deep VRegulome, a deep-learning method for prediction and interpretation of functionally disruptive variants in the human regulome, which combines 700 DNABERT fine-tuned models, trained on vast amounts of ENCODE gene regulatory regions, with variant scoring, motif analysis, attention-based visualization, and survival analysis. We showcase its application on TCGA glioblastoma WGS dataset in prioritizing survival-associated mutations and regulatory regions. The analysis identified 572 splice-disrupting and 9,837 transcription-factor binding site altering mutations occurring in greater than 10% of glioblastoma samples. Survival analysis linked 1352 mutations and 563 disrupted regulatory regions to patient outcomes, enabling stratification via non-coding mutation signatures. All the code, fine-tuned models, and an interactive data portal are publicly available.
MERGE: Multi-faceted Hierarchical Graph-based GNN for Gene Expression Prediction from Whole Slide Histopathology Images
Dec 03, 2024



Recent advances in Spatial Transcriptomics (ST) pair histology images with spatially resolved gene expression profiles, enabling predictions of gene expression across different tissue locations based on image patches. This opens up new possibilities for enhancing whole slide image (WSI) prediction tasks with localized gene expression. However, existing methods fail to fully leverage the interactions between different tissue locations, which are crucial for accurate joint prediction. To address this, we introduce MERGE (Multi-faceted hiErarchical gRaph for Gene Expressions), which combines a multi-faceted hierarchical graph construction strategy with graph neural networks (GNN) to improve gene expression predictions from WSIs. By clustering tissue image patches based on both spatial and morphological features, and incorporating intra- and inter-cluster edges, our approach fosters interactions between distant tissue locations during GNN learning. As an additional contribution, we evaluate different data smoothing techniques that are necessary to mitigate artifacts in ST data, often caused by technical imperfections. We advocate for adopting gene-aware smoothing methods that are more biologically justified. Experimental results on gene expression prediction show that our GNN method outperforms state-of-the-art techniques across multiple metrics.
RankByGene: Gene-Guided Histopathology Representation Learning Through Cross-Modal Ranking Consistency
Nov 22, 2024Spatial transcriptomics (ST) provides essential spatial context by mapping gene expression within tissue, enabling detailed study of cellular heterogeneity and tissue organization. However, aligning ST data with histology images poses challenges due to inherent spatial distortions and modality-specific variations. Existing methods largely rely on direct alignment, which often fails to capture complex cross-modal relationships. To address these limitations, we propose a novel framework that aligns gene and image features using a ranking-based alignment loss, preserving relative similarity across modalities and enabling robust multi-scale alignment. To further enhance the alignment's stability, we employ self-supervised knowledge distillation with a teacher-student network architecture, effectively mitigating disruptions from high dimensionality, sparsity, and noise in gene expression data. Extensive experiments on gene expression prediction and survival analysis demonstrate our framework's effectiveness, showing improved alignment and predictive performance over existing methods and establishing a robust tool for gene-guided image representation learning in digital pathology.