 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLarge Language Models with Retrieval-Augmented Generation for Zero-Shot Disease Phenotyping
Dec 11, 2023Identifying disease phenotypes from electronic health records (EHRs) is critical for numerous secondary uses. Manually encoding physician knowledge into rules is particularly challenging for rare diseases due to inadequate EHR coding, necessitating review of clinical notes. Large language models (LLMs) offer promise in text understanding but may not efficiently handle real-world clinical documentation. We propose a zero-shot LLM-based method enriched by retrieval-augmented generation and MapReduce, which pre-identifies disease-related text snippets to be used in parallel as queries for the LLM to establish diagnosis. We show that this method as applied to pulmonary hypertension (PH), a rare disease characterized by elevated arterial pressures in the lungs, significantly outperforms physician logic rules ($F_1$ score of 0.62 vs. 0.75). This method has the potential to enhance rare disease cohort identification, expanding the scope of robust clinical research and care gap identification.
Active learning for medical code assignment
Apr 12, 2021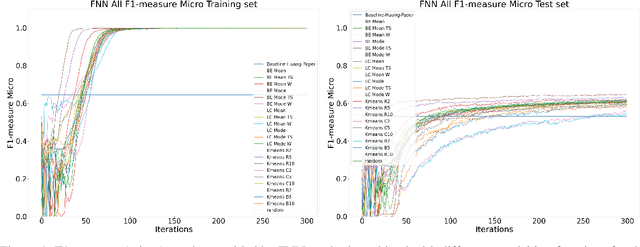
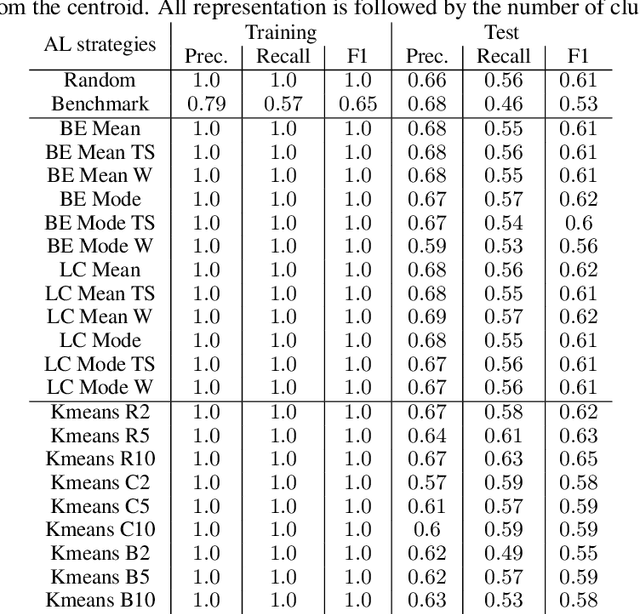
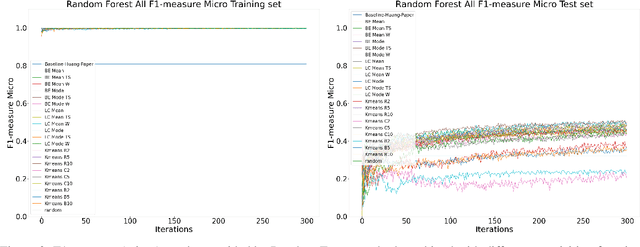
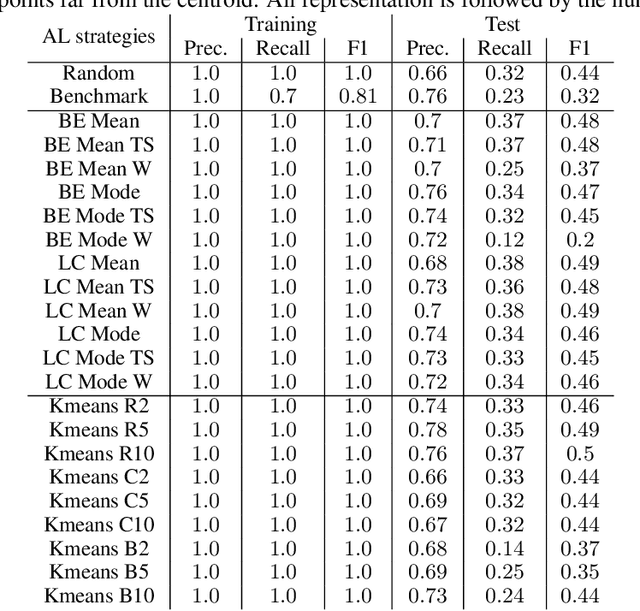
Machine Learning (ML) is widely used to automatically extract meaningful information from Electronic Health Records (EHR) to support operational, clinical, and financial decision-making. However, ML models require a large number of annotated examples to provide satisfactory results, which is not possible in most healthcare scenarios due to the high cost of clinician-labeled data. Active Learning (AL) is a process of selecting the most informative instances to be labeled by an expert to further train a supervised algorithm. We demonstrate the effectiveness of AL in multi-label text classification in the clinical domain. In this context, we apply a set of well-known AL methods to help automatically assign ICD-9 codes on the MIMIC-III dataset. Our results show that the selection of informative instances provides satisfactory classification with a significantly reduced training set (8.3\% of the total instances). We conclude that AL methods can significantly reduce the manual annotation cost while preserving model performance.
Contrastive Learning Improves Critical Event Prediction in COVID-19 Patients
Jan 11, 2021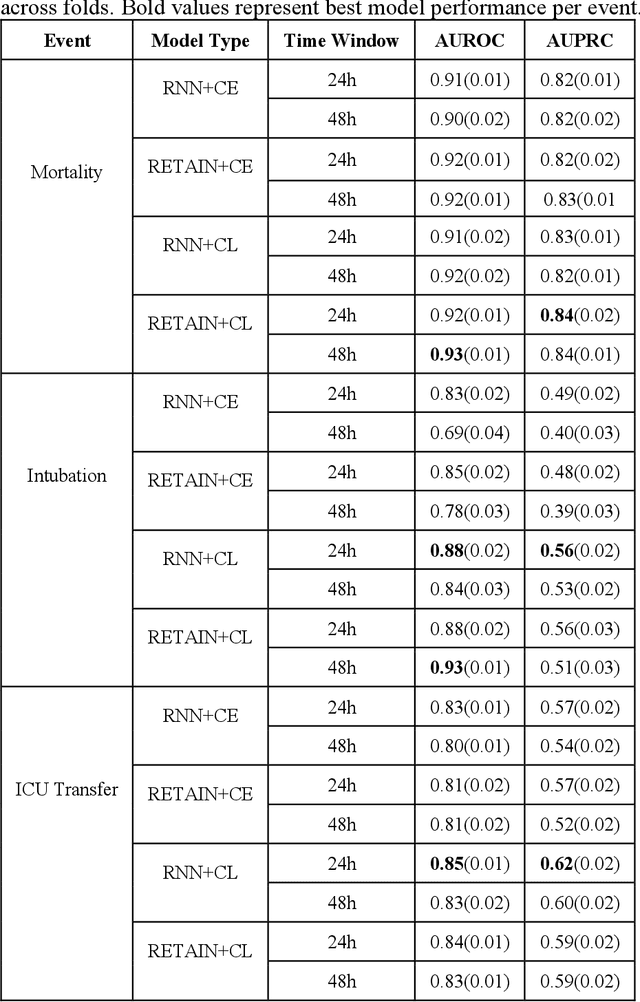
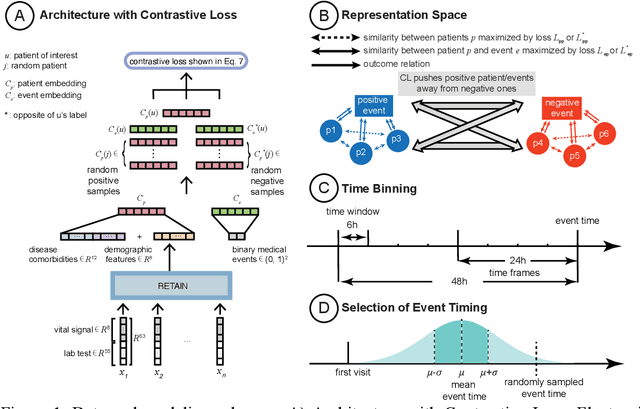
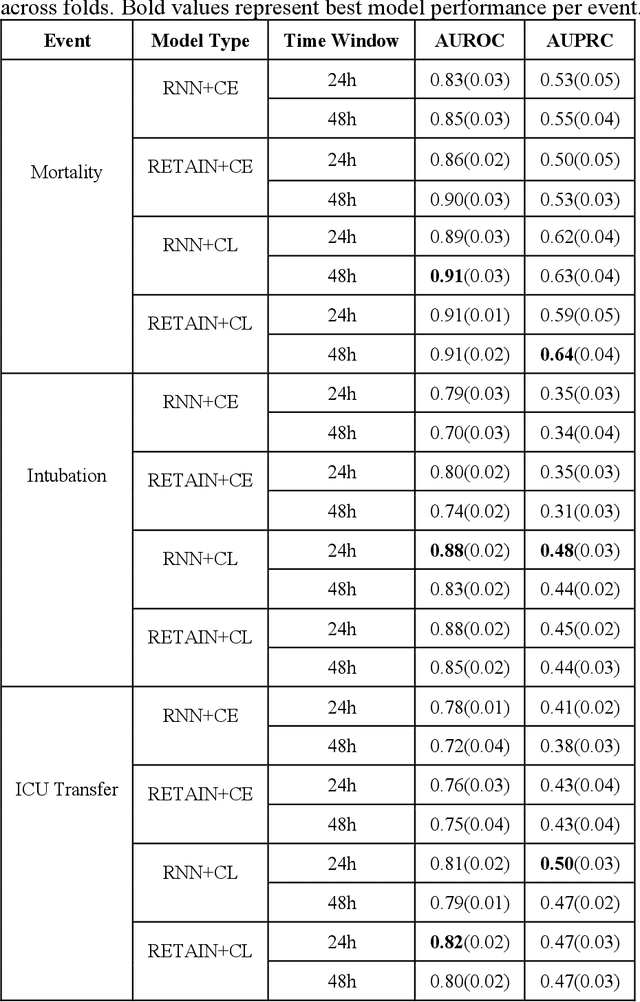
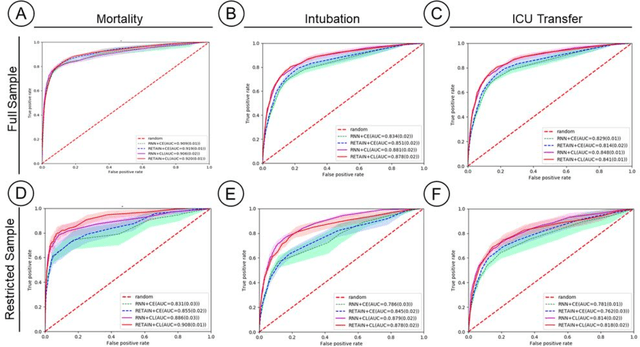
Machine Learning (ML) models typically require large-scale, balanced training data to be robust, generalizable, and effective in the context of healthcare. This has been a major issue for developing ML models for the coronavirus-disease 2019 (COVID-19) pandemic where data is highly imbalanced, particularly within electronic health records (EHR) research. Conventional approaches in ML use cross-entropy loss (CEL) that often suffers from poor margin classification. For the first time, we show that contrastive loss (CL) improves the performance of CEL especially for imbalanced EHR data and the related COVID-19 analyses. This study has been approved by the Institutional Review Board at the Icahn School of Medicine at Mount Sinai. We use EHR data from five hospitals within the Mount Sinai Health System (MSHS) to predict mortality, intubation, and intensive care unit (ICU) transfer in hospitalized COVID-19 patients over 24 and 48 hour time windows. We train two sequential architectures (RNN and RETAIN) using two loss functions (CEL and CL). Models are tested on full sample data set which contain all available data and restricted data set to emulate higher class imbalance.CL models consistently outperform CEL models with the restricted data set on these tasks with differences ranging from 0.04 to 0.15 for AUPRC and 0.05 to 0.1 for AUROC. For the restricted sample, only the CL model maintains proper clustering and is able to identify important features, such as pulse oximetry. CL outperforms CEL in instances of severe class imbalance, on three EHR outcomes with respect to three performance metrics: predictive power, clustering, and feature importance. We believe that the developed CL framework can be expanded and used for EHR ML work in general.
Deep Representation Learning of Electronic Health Records to Unlock Patient Stratification at Scale
Mar 14, 2020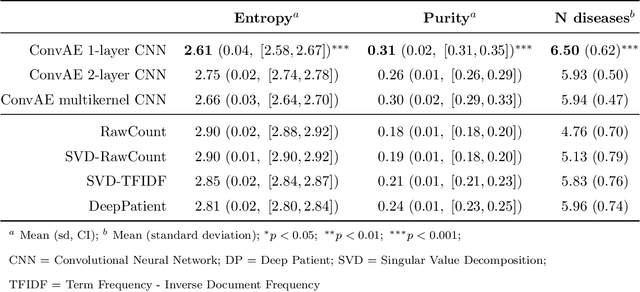
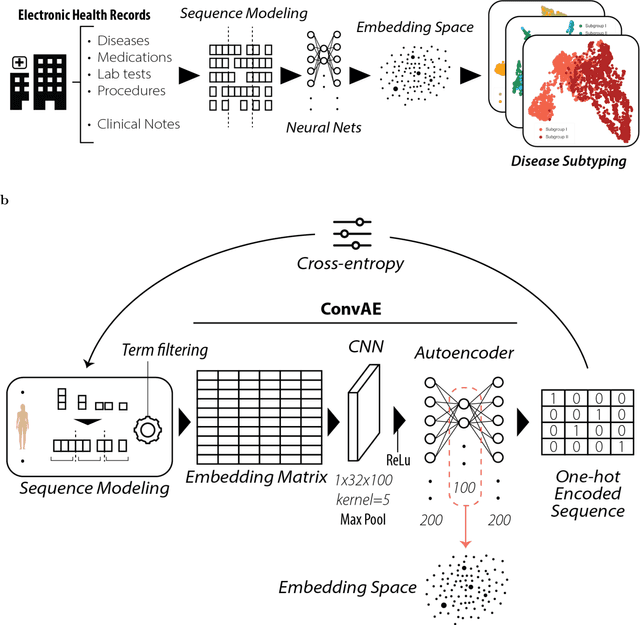
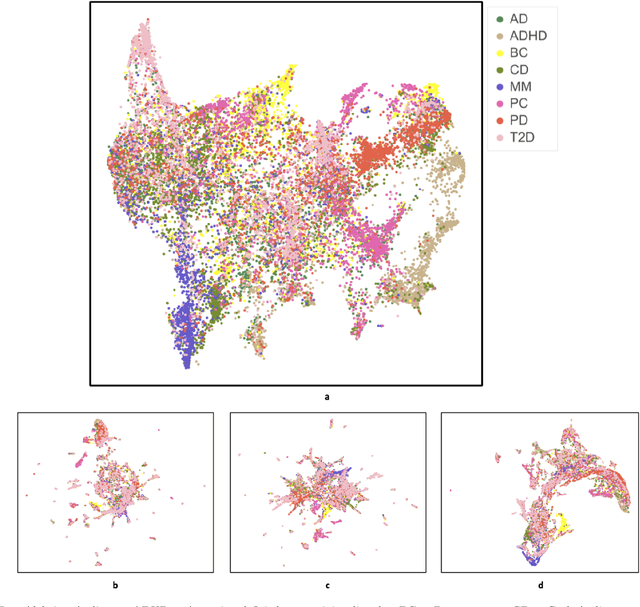
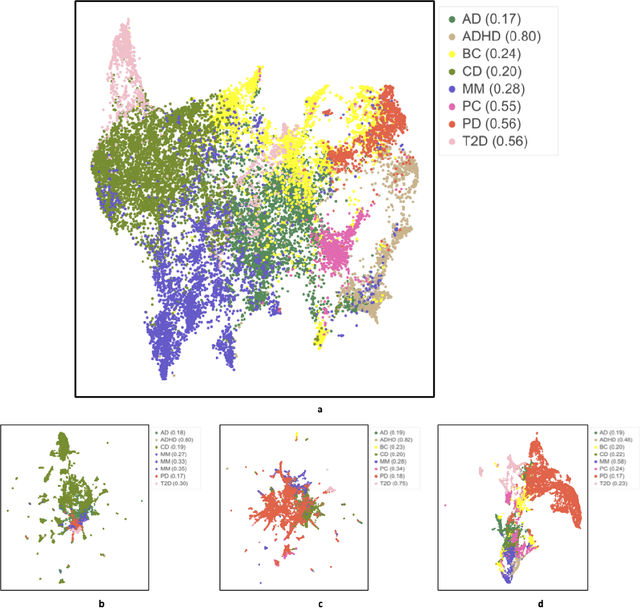
Objective: Deriving disease subtypes from electronic health records (EHRs) can guide next-generation personalized medicine. However, challenges in summarizing and representing patient data prevent widespread practice of scalable EHR-based stratification analysis. Here, we present a novel unsupervised framework based on deep learning to process heterogeneous EHRs and derive patient representations that can efficiently and effectively enable patient stratification at scale. Materials and methods: We considered EHRs of $1,608,741$ patients from a diverse hospital cohort comprising of a total of $57,464$ clinical concepts. We introduce a representation learning model based on word embeddings, convolutional neural networks and autoencoders (i.e., "ConvAE") to transform patient trajectories into low-dimensional latent vectors. We evaluated these representations as broadly enabling patient stratification by applying hierarchical clustering to different multi-disease and disease-specific patient cohorts. Results: ConvAE significantly outperformed several common baselines in a clustering task to identify patients with different complex conditions, with $2.61$ entropy and $0.31$ purity average scores. When applied to stratify patients within a certain condition, ConvAE led to various clinically relevant subtypes for different disorders, including type 2 diabetes, Parkinson's disease and Alzheimer's disease, largely related to comorbidities, disease progression, and symptom severity. Conclusions: Patient representations derived from modeling EHRs with ConvAE can help develop personalized medicine therapeutic strategies and better understand varying etiologies in heterogeneous sub-populations.
Scaling structural learning with NO-BEARS to infer causal transcriptome networks
Oct 31, 2019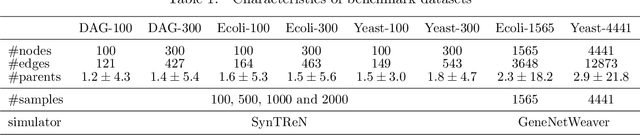

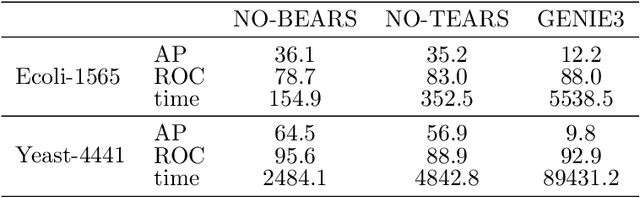
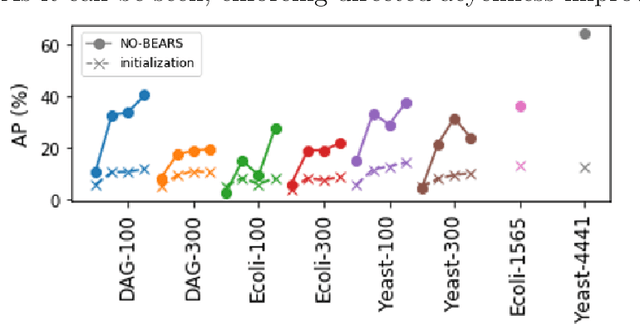
Constructing gene regulatory networks is a critical step in revealing disease mechanisms from transcriptomic data. In this work, we present NO-BEARS, a novel algorithm for estimating gene regulatory networks. The NO-BEARS algorithm is built on the basis of the NOTEARS algorithm with two improvements. First, we propose a new constraint and its fast approximation to reduce the computational cost of the NO-TEARS algorithm. Next, we introduce a polynomial regression loss to handle non-linearity in gene expressions. Our implementation utilizes modern GPU computation that can decrease the time of hours-long CPU computation to seconds. Using synthetic data, we demonstrate improved performance, both in processing time and accuracy, on inferring gene regulatory networks from gene expression data.
Enhancing high-content imaging for studying microtubule networks at large-scale
Oct 01, 2019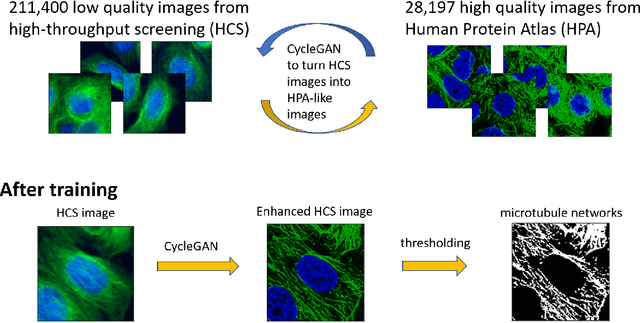
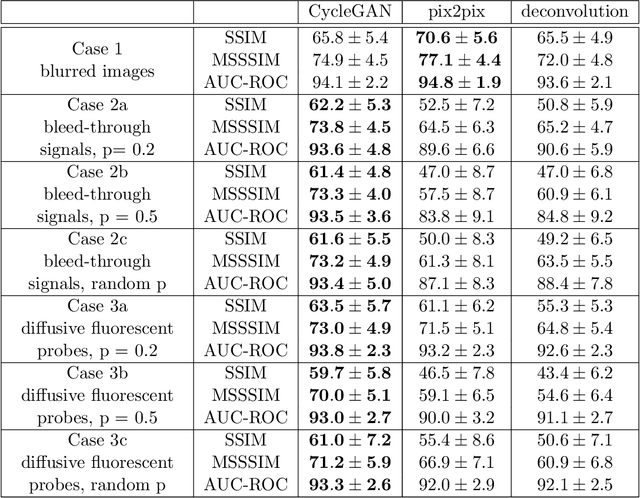
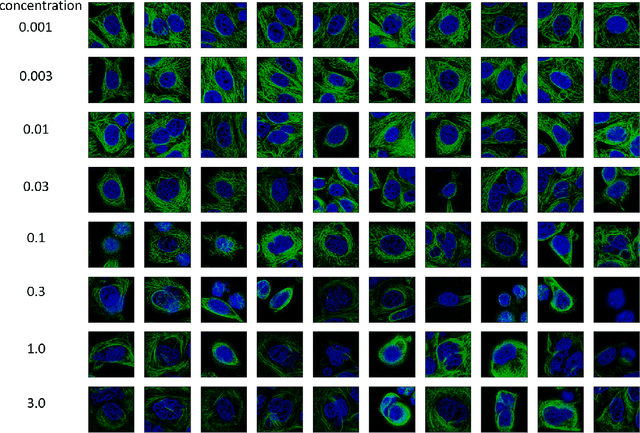
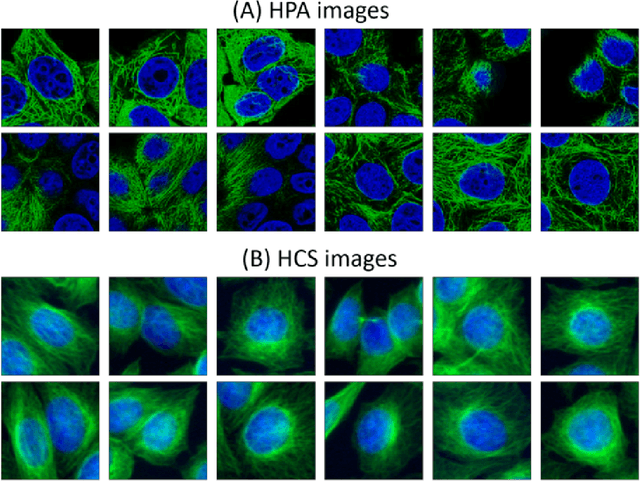
Given the crucial role of microtubules for cell survival, many researchers have found success using microtubule-targeting agents in the search for effective cancer therapeutics. Understanding microtubule responses to targeted interventions requires that the microtubule network within cells can be consistently observed across a large sample of images. However, fluorescence noise sources captured simultaneously with biological signals while using wide-field microscopes can obfuscate fine microtubule structures. Such requirements are particularly challenging for high-throughput imaging, where researchers must make decisions related to the trade-off between imaging quality and speed. Here, we propose a computational framework to enhance the quality of high-throughput imaging data to achieve fast speed and high quality simultaneously. Using CycleGAN, we learn an image model from low-throughput, high-resolution images to enhance features, such as microtubule networks in high-throughput low-resolution images. We show that CycleGAN is effective in identifying microtubules with 0.93+ AUC-ROC and that these results are robust to different kinds of image noise. We further apply CycleGAN to quantify the changes in microtubule density as a result of the application of drug compounds, and show that the quantified responses correspond well with known drug effects
Natural Language Processing of Clinical Notes on Chronic Diseases: Systematic Review
Aug 15, 2019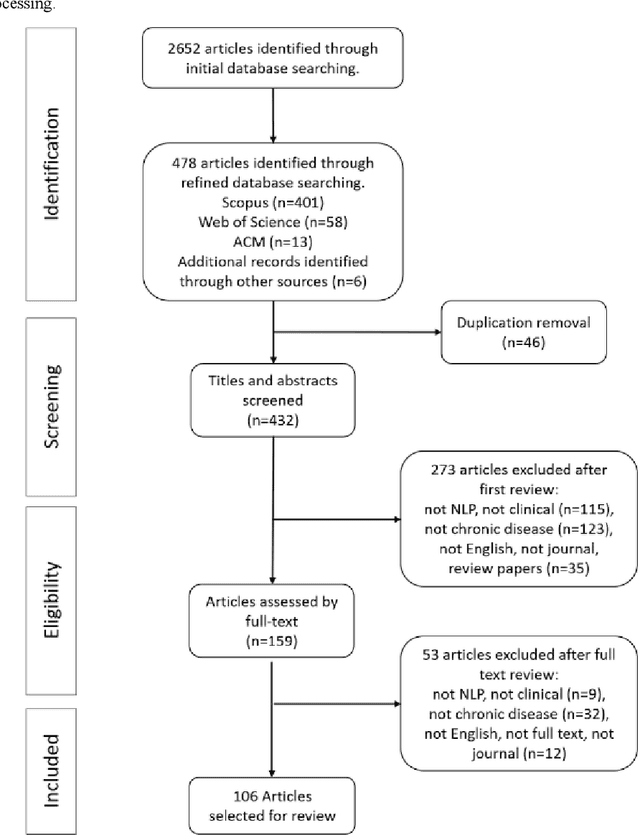

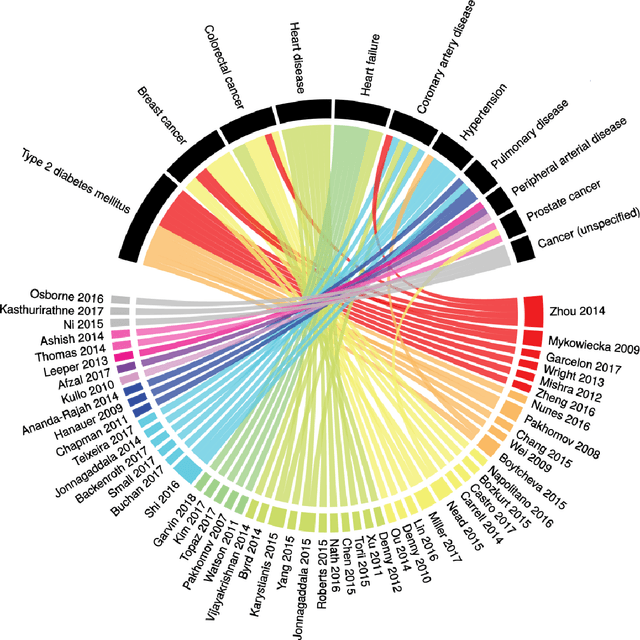

Of the 2652 articles considered, 106 met the inclusion criteria. Review of the included papers resulted in identification of 43 chronic diseases, which were then further classified into 10 disease categories using ICD-10. The majority of studies focused on diseases of the circulatory system (n=38) while endocrine and metabolic diseases were fewest (n=14). This was due to the structure of clinical records related to metabolic diseases, which typically contain much more structured data, compared with medical records for diseases of the circulatory system, which focus more on unstructured data and consequently have seen a stronger focus of NLP. The review has shown that there is a significant increase in the use of machine learning methods compared to rule-based approaches; however, deep learning methods remain emergent (n=3). Consequently, the majority of works focus on classification of disease phenotype with only a handful of papers addressing extraction of comorbidities from the free text or integration of clinical notes with structured data. There is a notable use of relatively simple methods, such as shallow classifiers (or combination with rule-based methods), due to the interpretability of predictions, which still represents a significant issue for more complex methods. Finally, scarcity of publicly available data may also have contributed to insufficient development of more advanced methods, such as extraction of word embeddings from clinical notes. Further efforts are still required to improve (1) progression of clinical NLP methods from extraction toward understanding; (2) recognition of relations among entities rather than entities in isolation; (3) temporal extraction to understand past, current, and future clinical events; (4) exploitation of alternative sources of clinical knowledge; and (5) availability of large-scale, de-identified clinical corpora.
* Supplementary material detailing articles reviewed, classification of diseases and associated algorithms, can be found at: http://venetosmani.com/research/publications.html