 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDeep Representation Learning of Electronic Health Records to Unlock Patient Stratification at Scale
Mar 14, 2020
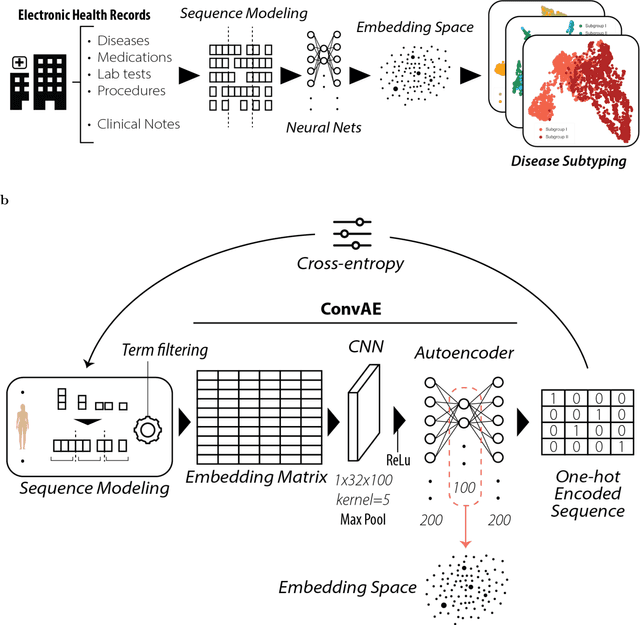
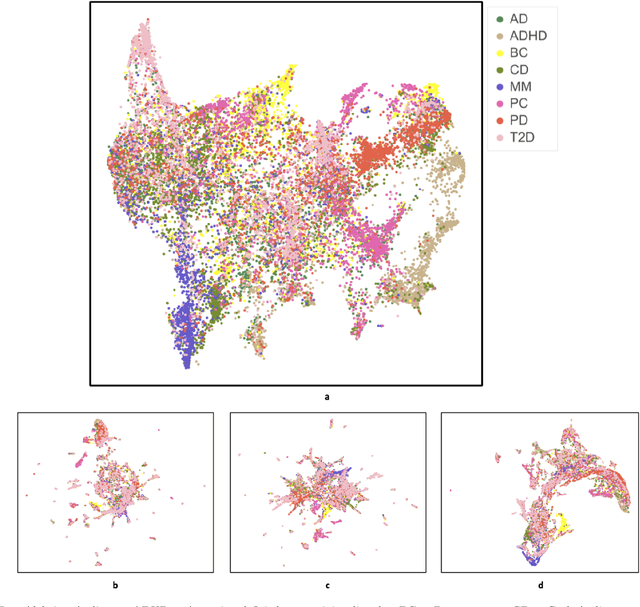
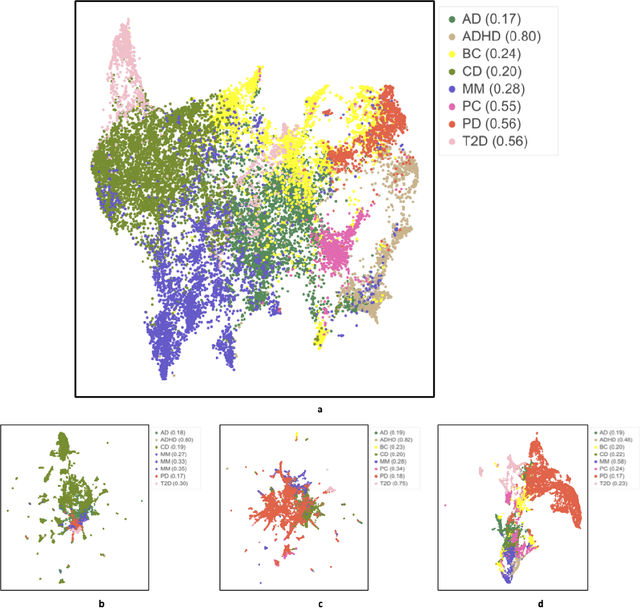
Objective: Deriving disease subtypes from electronic health records (EHRs) can guide next-generation personalized medicine. However, challenges in summarizing and representing patient data prevent widespread practice of scalable EHR-based stratification analysis. Here, we present a novel unsupervised framework based on deep learning to process heterogeneous EHRs and derive patient representations that can efficiently and effectively enable patient stratification at scale. Materials and methods: We considered EHRs of $1,608,741$ patients from a diverse hospital cohort comprising of a total of $57,464$ clinical concepts. We introduce a representation learning model based on word embeddings, convolutional neural networks and autoencoders (i.e., "ConvAE") to transform patient trajectories into low-dimensional latent vectors. We evaluated these representations as broadly enabling patient stratification by applying hierarchical clustering to different multi-disease and disease-specific patient cohorts. Results: ConvAE significantly outperformed several common baselines in a clustering task to identify patients with different complex conditions, with $2.61$ entropy and $0.31$ purity average scores. When applied to stratify patients within a certain condition, ConvAE led to various clinically relevant subtypes for different disorders, including type 2 diabetes, Parkinson's disease and Alzheimer's disease, largely related to comorbidities, disease progression, and symptom severity. Conclusions: Patient representations derived from modeling EHRs with ConvAE can help develop personalized medicine therapeutic strategies and better understand varying etiologies in heterogeneous sub-populations.
Scaling structural learning with NO-BEARS to infer causal transcriptome networks
Oct 31, 2019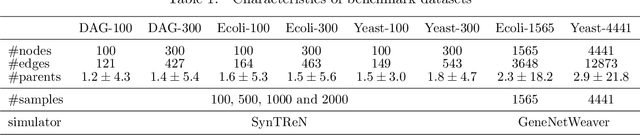

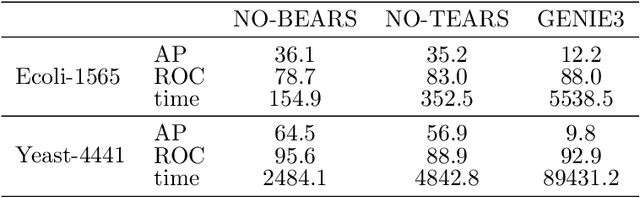
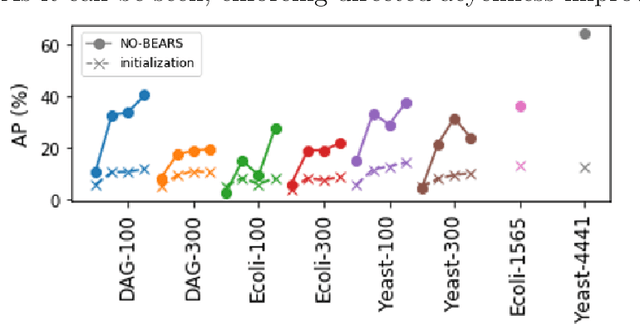
Constructing gene regulatory networks is a critical step in revealing disease mechanisms from transcriptomic data. In this work, we present NO-BEARS, a novel algorithm for estimating gene regulatory networks. The NO-BEARS algorithm is built on the basis of the NOTEARS algorithm with two improvements. First, we propose a new constraint and its fast approximation to reduce the computational cost of the NO-TEARS algorithm. Next, we introduce a polynomial regression loss to handle non-linearity in gene expressions. Our implementation utilizes modern GPU computation that can decrease the time of hours-long CPU computation to seconds. Using synthetic data, we demonstrate improved performance, both in processing time and accuracy, on inferring gene regulatory networks from gene expression data.