 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOpen-H-Embodiment: A Large-Scale Dataset for Enabling Foundation Models in Medical Robotics
Apr 22, 2026Autonomous medical robots hold promise to improve patient outcomes, reduce provider workload, democratize access to care, and enable superhuman precision. However, autonomous medical robotics has been limited by a fundamental data problem: existing medical robotic datasets are small, single-embodiment, and rarely shared openly, restricting the development of foundation models that the field needs to advance. We introduce Open-H-Embodiment, the largest open dataset of medical robotic video with synchronized kinematics to date, spanning more than 49 institutions and multiple robotic platforms including the CMR Versius, Intuitive Surgical's da Vinci, da Vinci Research Kit (dVRK), Rob Surgical BiTrack, Virtual Incision's MIRA, Moon Surgical Maestro, and a variety of custom systems, spanning surgical manipulation, robotic ultrasound, and endoscopy procedures. We demonstrate the research enabled by this dataset through two foundation models. GR00T-H is the first open foundation vision-language-action model for medical robotics, which is the only evaluated model to achieve full end-to-end task completion on a structured suturing benchmark (25% of trials vs. 0% for all others) and achieves 64% average success across a 29-step ex vivo suturing sequence. We also train Cosmos-H-Surgical-Simulator, the first action-conditioned world model to enable multi-embodiment surgical simulation from a single checkpoint, spanning nine robotic platforms and supporting in silico policy evaluation and synthetic data generation for the medical domain. These results suggest that open, large-scale medical robot data collection can serve as critical infrastructure for the research community, enabling advances in robot learning, world modeling, and beyond.
Deep Representation Learning of Electronic Health Records to Unlock Patient Stratification at Scale
Mar 14, 2020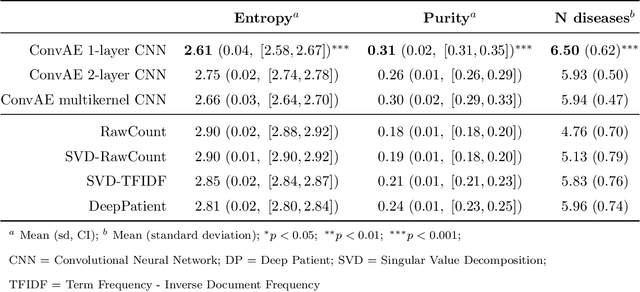
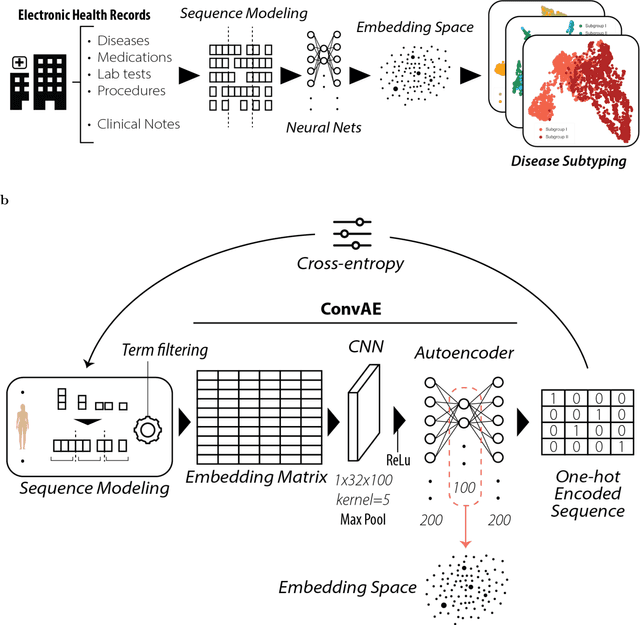
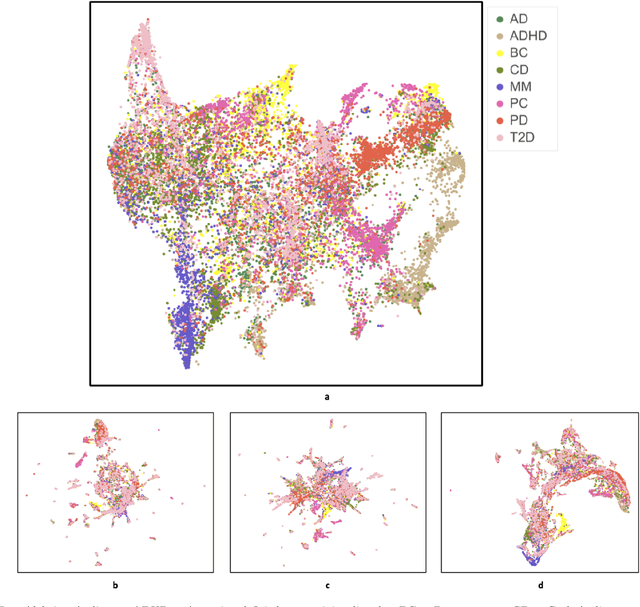
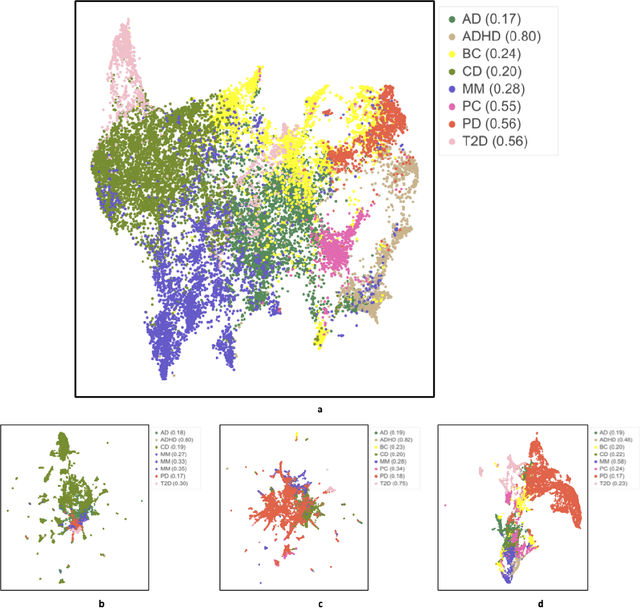
Objective: Deriving disease subtypes from electronic health records (EHRs) can guide next-generation personalized medicine. However, challenges in summarizing and representing patient data prevent widespread practice of scalable EHR-based stratification analysis. Here, we present a novel unsupervised framework based on deep learning to process heterogeneous EHRs and derive patient representations that can efficiently and effectively enable patient stratification at scale. Materials and methods: We considered EHRs of $1,608,741$ patients from a diverse hospital cohort comprising of a total of $57,464$ clinical concepts. We introduce a representation learning model based on word embeddings, convolutional neural networks and autoencoders (i.e., "ConvAE") to transform patient trajectories into low-dimensional latent vectors. We evaluated these representations as broadly enabling patient stratification by applying hierarchical clustering to different multi-disease and disease-specific patient cohorts. Results: ConvAE significantly outperformed several common baselines in a clustering task to identify patients with different complex conditions, with $2.61$ entropy and $0.31$ purity average scores. When applied to stratify patients within a certain condition, ConvAE led to various clinically relevant subtypes for different disorders, including type 2 diabetes, Parkinson's disease and Alzheimer's disease, largely related to comorbidities, disease progression, and symptom severity. Conclusions: Patient representations derived from modeling EHRs with ConvAE can help develop personalized medicine therapeutic strategies and better understand varying etiologies in heterogeneous sub-populations.
Scaling structural learning with NO-BEARS to infer causal transcriptome networks
Oct 31, 2019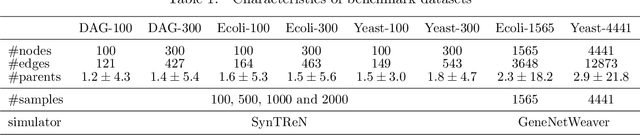

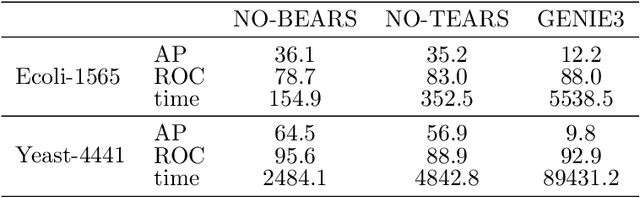
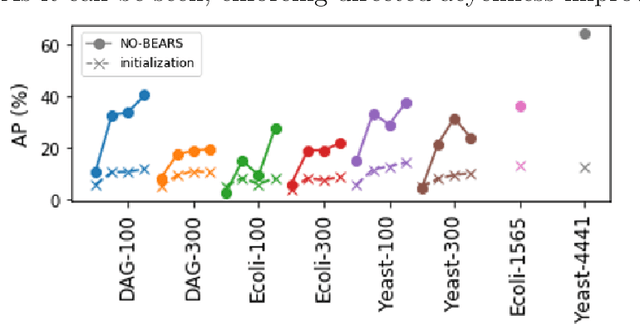
Constructing gene regulatory networks is a critical step in revealing disease mechanisms from transcriptomic data. In this work, we present NO-BEARS, a novel algorithm for estimating gene regulatory networks. The NO-BEARS algorithm is built on the basis of the NOTEARS algorithm with two improvements. First, we propose a new constraint and its fast approximation to reduce the computational cost of the NO-TEARS algorithm. Next, we introduce a polynomial regression loss to handle non-linearity in gene expressions. Our implementation utilizes modern GPU computation that can decrease the time of hours-long CPU computation to seconds. Using synthetic data, we demonstrate improved performance, both in processing time and accuracy, on inferring gene regulatory networks from gene expression data.
Enhancing high-content imaging for studying microtubule networks at large-scale
Oct 01, 2019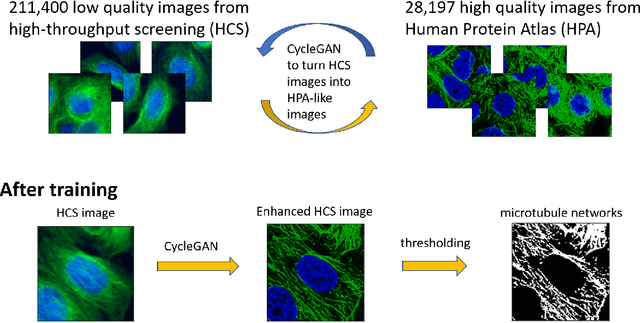
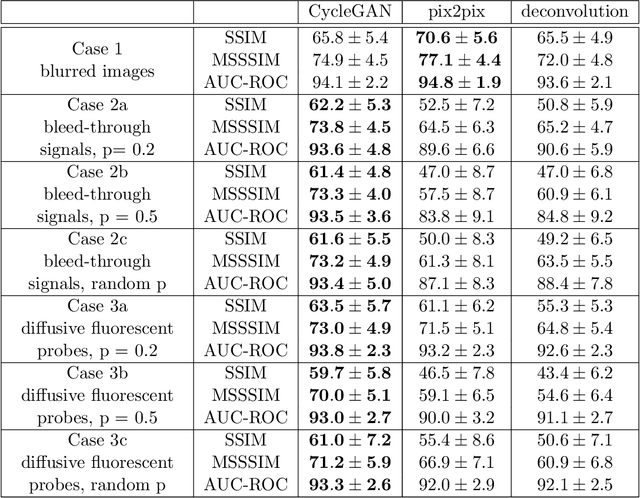
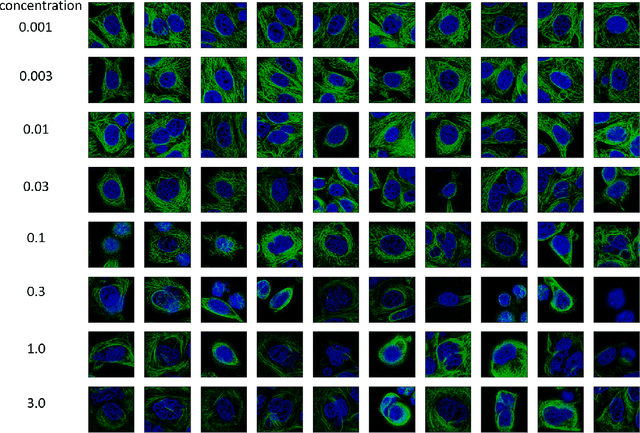
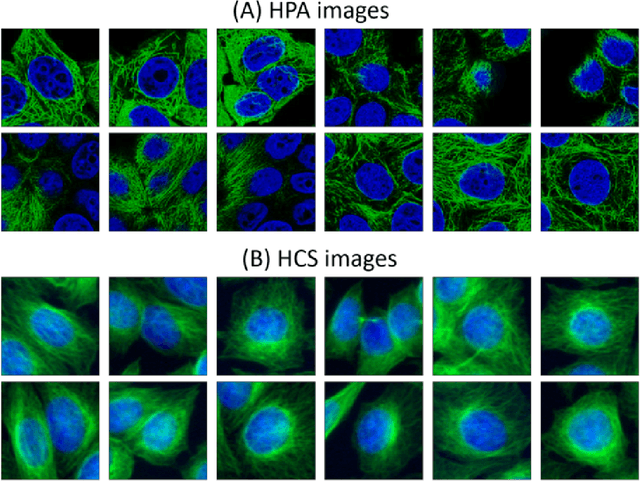
Given the crucial role of microtubules for cell survival, many researchers have found success using microtubule-targeting agents in the search for effective cancer therapeutics. Understanding microtubule responses to targeted interventions requires that the microtubule network within cells can be consistently observed across a large sample of images. However, fluorescence noise sources captured simultaneously with biological signals while using wide-field microscopes can obfuscate fine microtubule structures. Such requirements are particularly challenging for high-throughput imaging, where researchers must make decisions related to the trade-off between imaging quality and speed. Here, we propose a computational framework to enhance the quality of high-throughput imaging data to achieve fast speed and high quality simultaneously. Using CycleGAN, we learn an image model from low-throughput, high-resolution images to enhance features, such as microtubule networks in high-throughput low-resolution images. We show that CycleGAN is effective in identifying microtubules with 0.93+ AUC-ROC and that these results are robust to different kinds of image noise. We further apply CycleGAN to quantify the changes in microtubule density as a result of the application of drug compounds, and show that the quantified responses correspond well with known drug effects