 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeExtracting Post-Acute Sequelae of SARS-CoV-2 Infection Symptoms from Clinical Notes via Hybrid Natural Language Processing
Aug 17, 2025Accurately and efficiently diagnosing Post-Acute Sequelae of COVID-19 (PASC) remains challenging due to its myriad symptoms that evolve over long- and variable-time intervals. To address this issue, we developed a hybrid natural language processing pipeline that integrates rule-based named entity recognition with BERT-based assertion detection modules for PASC-symptom extraction and assertion detection from clinical notes. We developed a comprehensive PASC lexicon with clinical specialists. From 11 health systems of the RECOVER initiative network across the U.S., we curated 160 intake progress notes for model development and evaluation, and collected 47,654 progress notes for a population-level prevalence study. We achieved an average F1 score of 0.82 in one-site internal validation and 0.76 in 10-site external validation for assertion detection. Our pipeline processed each note at $2.448\pm 0.812$ seconds on average. Spearman correlation tests showed $\rho >0.83$ for positive mentions and $\rho >0.72$ for negative ones, both with $P <0.0001$. These demonstrate the effectiveness and efficiency of our models and their potential for improving PASC diagnosis.
Federated Causal Inference in Healthcare: Methods, Challenges, and Applications
May 04, 2025



Federated causal inference enables multi-site treatment effect estimation without sharing individual-level data, offering a privacy-preserving solution for real-world evidence generation. However, data heterogeneity across sites, manifested in differences in covariate, treatment, and outcome, poses significant challenges for unbiased and efficient estimation. In this paper, we present a comprehensive review and theoretical analysis of federated causal effect estimation across both binary/continuous and time-to-event outcomes. We classify existing methods into weight-based strategies and optimization-based frameworks and further discuss extensions including personalized models, peer-to-peer communication, and model decomposition. For time-to-event outcomes, we examine federated Cox and Aalen-Johansen models, deriving asymptotic bias and variance under heterogeneity. Our analysis reveals that FedProx-style regularization achieves near-optimal bias-variance trade-offs compared to naive averaging and meta-analysis. We review related software tools and conclude by outlining opportunities, challenges, and future directions for scalable, fair, and trustworthy federated causal inference in distributed healthcare systems.
Emerging Opportunities of Using Large Language Models for Translation Between Drug Molecules and Indications
Feb 16, 2024A drug molecule is a substance that changes the organism's mental or physical state. Every approved drug has an indication, which refers to the therapeutic use of that drug for treating a particular medical condition. While the Large Language Model (LLM), a generative Artificial Intelligence (AI) technique, has recently demonstrated effectiveness in translating between molecules and their textual descriptions, there remains a gap in research regarding their application in facilitating the translation between drug molecules and indications, or vice versa, which could greatly benefit the drug discovery process. The capability of generating a drug from a given indication would allow for the discovery of drugs targeting specific diseases or targets and ultimately provide patients with better treatments. In this paper, we first propose a new task, which is the translation between drug molecules and corresponding indications, and then test existing LLMs on this new task. Specifically, we consider nine variations of the T5 LLM and evaluate them on two public datasets obtained from ChEMBL and DrugBank. Our experiments show the early results of using LLMs for this task and provide a perspective on the state-of-the-art. We also emphasize the current limitations and discuss future work that has the potential to improve the performance on this task. The creation of molecules from indications, or vice versa, will allow for more efficient targeting of diseases and significantly reduce the cost of drug discovery, with the potential to revolutionize the field of drug discovery in the era of generative AI.
SCEHR: Supervised Contrastive Learning for Clinical Risk Prediction using Electronic Health Records
Oct 11, 2021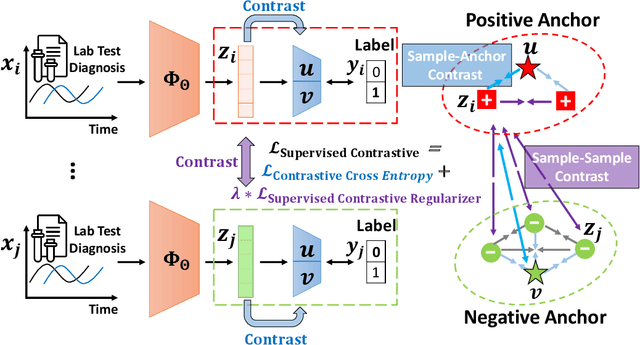
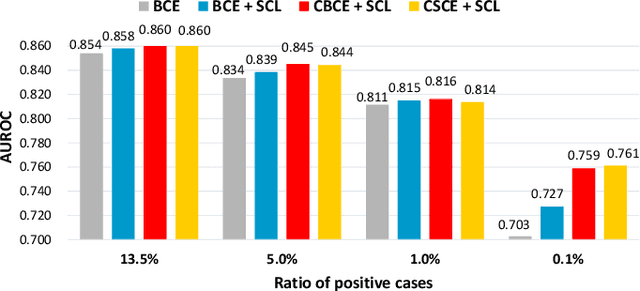

Contrastive learning has demonstrated promising performance in image and text domains either in a self-supervised or a supervised manner. In this work, we extend the supervised contrastive learning framework to clinical risk prediction problems based on longitudinal electronic health records (EHR). We propose a general supervised contrastive loss $\mathcal{L}_{\text{Contrastive Cross Entropy} } + \lambda \mathcal{L}_{\text{Supervised Contrastive Regularizer}}$ for learning both binary classification (e.g. in-hospital mortality prediction) and multi-label classification (e.g. phenotyping) in a unified framework. Our supervised contrastive loss practices the key idea of contrastive learning, namely, pulling similar samples closer and pushing dissimilar ones apart from each other, simultaneously by its two components: $\mathcal{L}_{\text{Contrastive Cross Entropy} }$ tries to contrast samples with learned anchors which represent positive and negative clusters, and $\mathcal{L}_{\text{Supervised Contrastive Regularizer}}$ tries to contrast samples with each other according to their supervised labels. We propose two versions of the above supervised contrastive loss and our experiments on real-world EHR data demonstrate that our proposed loss functions show benefits in improving the performance of strong baselines and even state-of-the-art models on benchmarking tasks for clinical risk predictions. Our loss functions work well with extremely imbalanced data which are common for clinical risk prediction problems. Our loss functions can be easily used to replace (binary or multi-label) cross-entropy loss adopted in existing clinical predictive models. The Pytorch code is released at \url{https://github.com/calvin-zcx/SCEHR}.
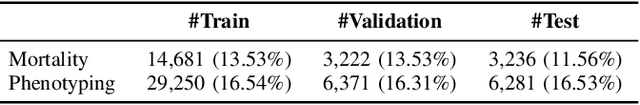
Contrastive Learning Improves Critical Event Prediction in COVID-19 Patients
Jan 11, 2021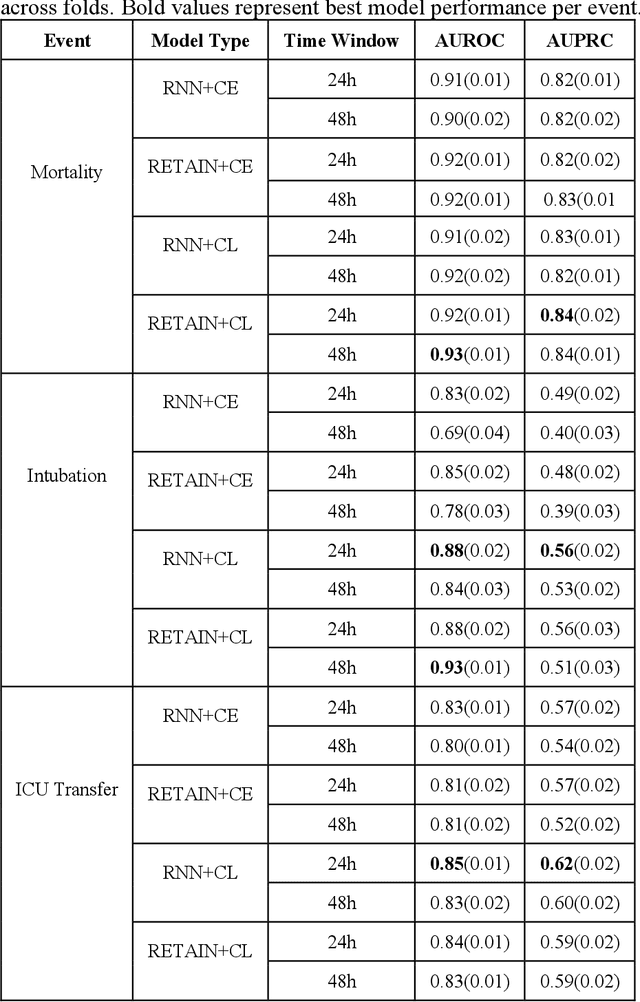
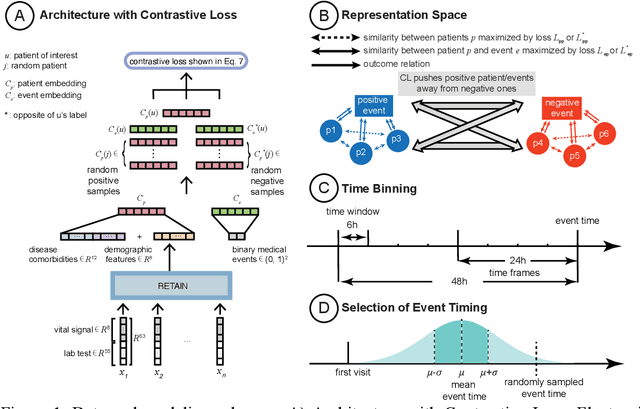
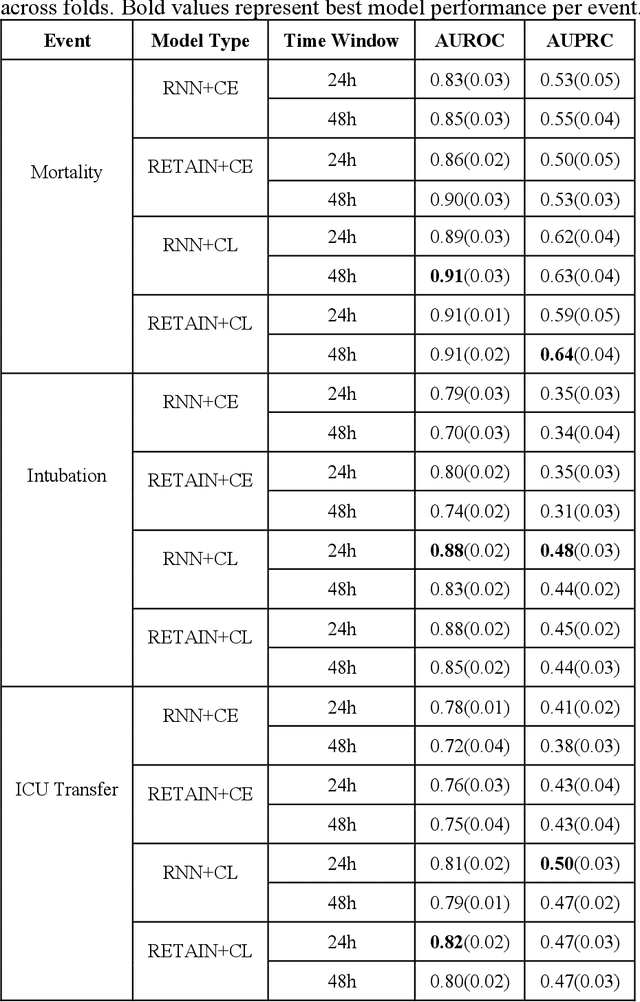
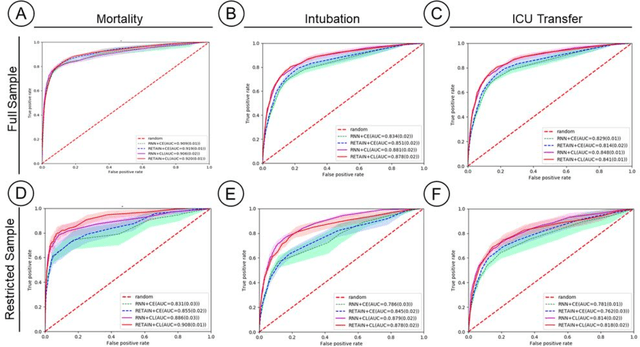
Machine Learning (ML) models typically require large-scale, balanced training data to be robust, generalizable, and effective in the context of healthcare. This has been a major issue for developing ML models for the coronavirus-disease 2019 (COVID-19) pandemic where data is highly imbalanced, particularly within electronic health records (EHR) research. Conventional approaches in ML use cross-entropy loss (CEL) that often suffers from poor margin classification. For the first time, we show that contrastive loss (CL) improves the performance of CEL especially for imbalanced EHR data and the related COVID-19 analyses. This study has been approved by the Institutional Review Board at the Icahn School of Medicine at Mount Sinai. We use EHR data from five hospitals within the Mount Sinai Health System (MSHS) to predict mortality, intubation, and intensive care unit (ICU) transfer in hospitalized COVID-19 patients over 24 and 48 hour time windows. We train two sequential architectures (RNN and RETAIN) using two loss functions (CEL and CL). Models are tested on full sample data set which contain all available data and restricted data set to emulate higher class imbalance.CL models consistently outperform CEL models with the restricted data set on these tasks with differences ranging from 0.04 to 0.15 for AUPRC and 0.05 to 0.1 for AUROC. For the restricted sample, only the CL model maintains proper clustering and is able to identify important features, such as pulse oximetry. CL outperforms CEL in instances of severe class imbalance, on three EHR outcomes with respect to three performance metrics: predictive power, clustering, and feature importance. We believe that the developed CL framework can be expanded and used for EHR ML work in general.
Visualizing Deep Graph Generative Models for Drug Discovery
Jul 20, 2020
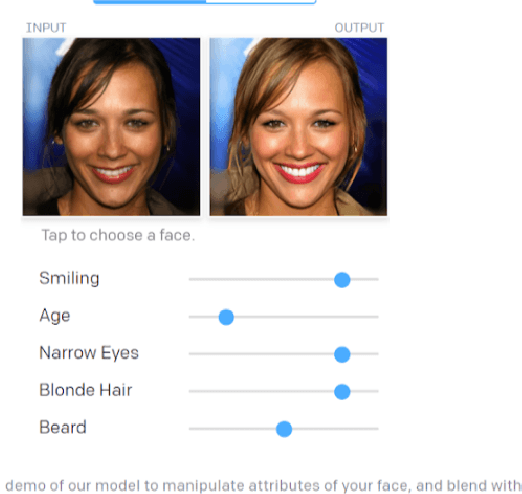
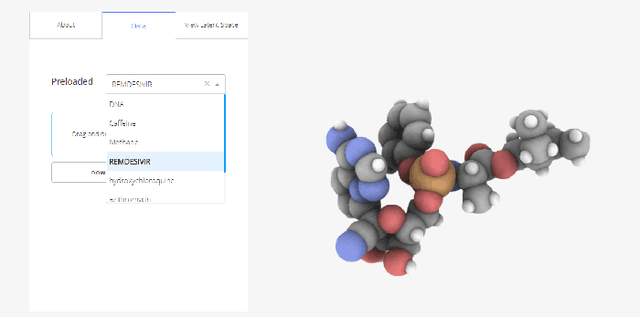
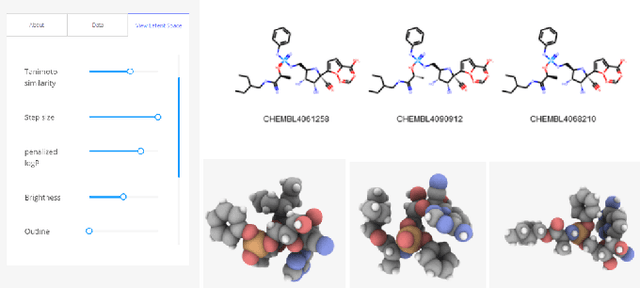
Drug discovery aims at designing novel molecules with specific desired properties for clinical trials. Over past decades, drug discovery and development have been a costly and time consuming process. Driven by big chemical data and AI, deep generative models show great potential to accelerate the drug discovery process. Existing works investigate different deep generative frameworks for molecular generation, however, less attention has been paid to the visualization tools to quickly demo and evaluate model's results. Here, we propose a visualization framework which provides interactive visualization tools to visualize molecules generated during the encoding and decoding process of deep graph generative models, and provide real time molecular optimization functionalities. Our work tries to empower black box AI driven drug discovery models with some visual interpretabilities.
MoFlow: An Invertible Flow Model for Generating Molecular Graphs
Jun 17, 2020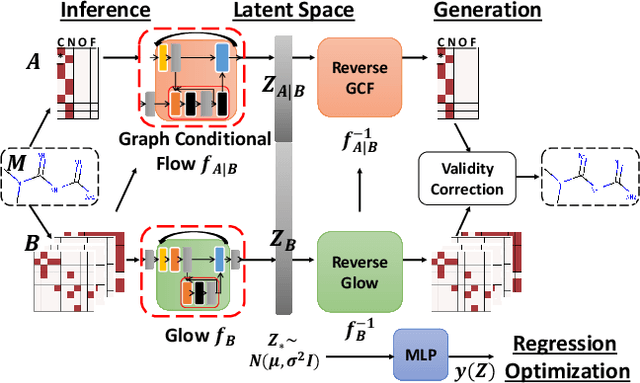
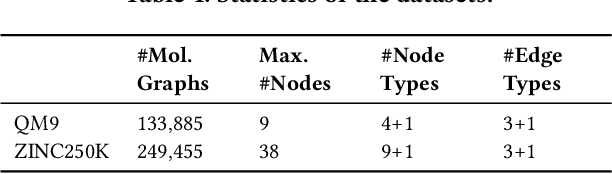
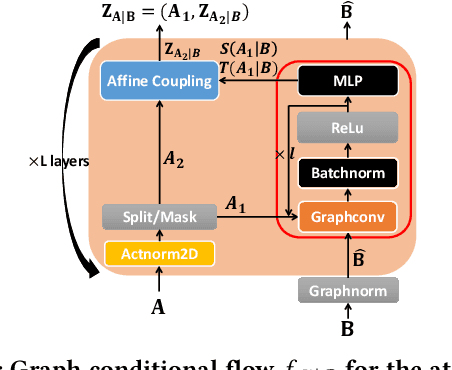
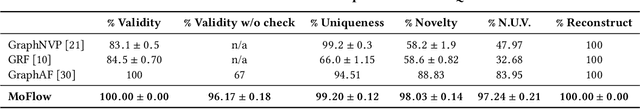
Generating molecular graphs with desired chemical properties driven by deep graph generative models provides a very promising way to accelerate drug discovery process. Such graph generative models usually consist of two steps: learning latent representations and generation of molecular graphs. However, to generate novel and chemically-valid molecular graphs from latent representations is very challenging because of the chemical constraints and combinatorial complexity of molecular graphs. In this paper, we propose MoFlow, a flow-based graph generative model to learn invertible mappings between molecular graphs and their latent representations. To generate molecular graphs, our MoFlow first generates bonds (edges) through a Glow based model, then generates atoms (nodes) given bonds by a novel graph conditional flow, and finally assembles them into a chemically valid molecular graph with a posthoc validity correction. Our MoFlow has merits including exact and tractable likelihood training, efficient one-pass embedding and generation, chemical validity guarantees, 100\% reconstruction of training data, and good generalization ability. We validate our model by four tasks: molecular graph generation and reconstruction, visualization of the continuous latent space, property optimization, and constrained property optimization. Our MoFlow achieves state-of-the-art performance, which implies its potential efficiency and effectiveness to explore large chemical space for drug discovery.
Neural Dynamics on Complex Networks
Aug 18, 2019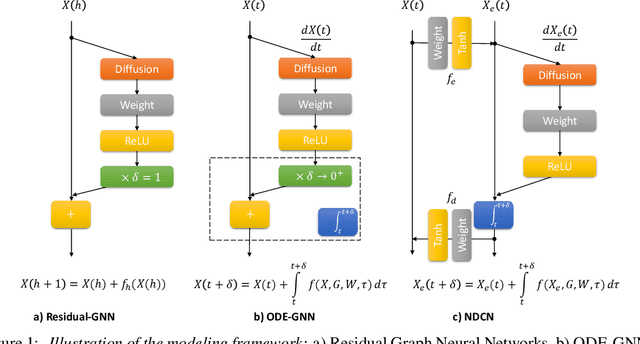
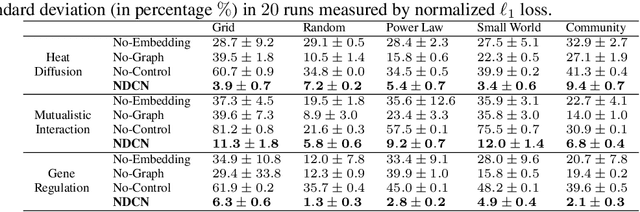
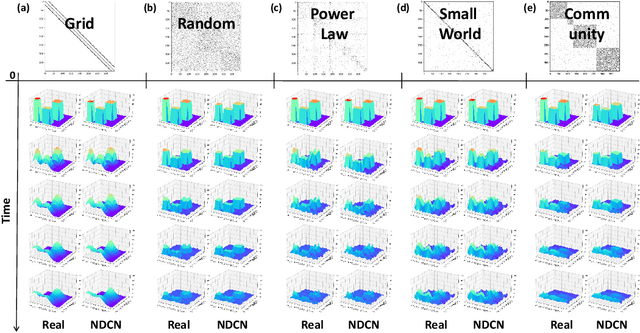
We introduce a deep learning model to learn continuous-time dynamics on complex networks and infer the semantic labels of nodes in the network at terminal time. We formulate the problem as an optimal control problem by minimizing a loss function consisting of a running loss of network dynamics, a terminal loss of nodes' labels, and a neural-differential-equation-system constraint. We solve the problem by a differential deep learning framework: as for the forward process of the system, rather than forwarding through a discrete number of hidden layers, we integrate the ordinary differential equation systems on graphs over continuous time; as for the backward learning process, we learn the optimal control parameters by back-propagation during solving initial value problem. We validate our model by learning complex dynamics on various real-world complex networks, and then apply our model to graph semi-supervised classification tasks. The promising experimental results demonstrate our model's capability of jointly capturing the structure, dynamics and semantics of complex systems.