 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNTIRE 2025 Challenge on Video Quality Enhancement for Video Conferencing: Datasets, Methods and Results
May 25, 2025This paper presents a comprehensive review of the 1st Challenge on Video Quality Enhancement for Video Conferencing held at the NTIRE workshop at CVPR 2025, and highlights the problem statement, datasets, proposed solutions, and results. The aim of this challenge was to design a Video Quality Enhancement (VQE) model to enhance video quality in video conferencing scenarios by (a) improving lighting, (b) enhancing colors, (c) reducing noise, and (d) enhancing sharpness - giving a professional studio-like effect. Participants were given a differentiable Video Quality Assessment (VQA) model, training, and test videos. A total of 91 participants registered for the challenge. We received 10 valid submissions that were evaluated in a crowdsourced framework.
Multiple Kronecker RLS fusion-based link propagation for drug-side effect prediction
Jun 27, 2024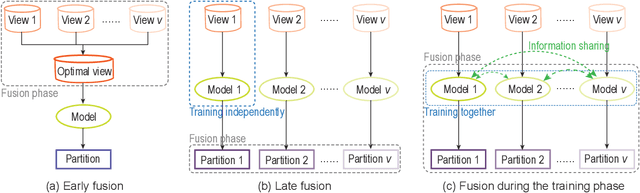
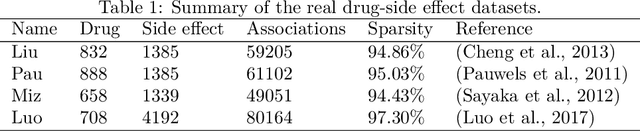
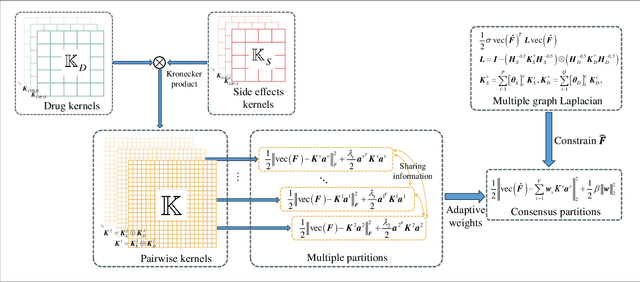
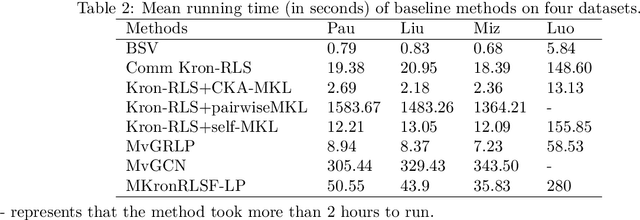
Drug-side effect prediction has become an essential area of research in the field of pharmacology. As the use of medications continues to rise, so does the importance of understanding and mitigating the potential risks associated with them. At present, researchers have turned to data-driven methods to predict drug-side effects. Drug-side effect prediction is a link prediction problem, and the related data can be described from various perspectives. To process these kinds of data, a multi-view method, called Multiple Kronecker RLS fusion-based link propagation (MKronRLSF-LP), is proposed. MKronRLSF-LP extends the Kron-RLS by finding the consensus partitions and multiple graph Laplacian constraints in the multi-view setting. Both of these multi-view settings contribute to a higher quality result. Extensive experiments have been conducted on drug-side effect datasets, and our empirical results provide evidence that our approach is effective and robust.
SBSM-Pro: Support Bio-sequence Machine for Proteins
Aug 20, 2023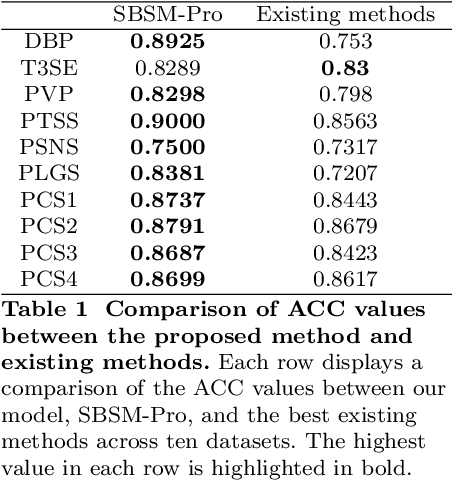



Proteins play a pivotal role in biological systems. The use of machine learning algorithms for protein classification can assist and even guide biological experiments, offering crucial insights for biotechnological applications. We propose a support bio-sequence machine for proteins, a model specifically designed for biological sequence classification. This model starts with raw sequences and groups amino acids based on their physicochemical properties. It incorporates sequence alignment to measure the similarities between proteins and uses a novel MKL approach to integrate various types of information, utilizing support vector machines for classification prediction. The results indicate that our model demonstrates commendable performance across 10 datasets in terms of the identification of protein function and posttranslational modification. This research not only showcases state-of-the-art work in protein classification but also paves the way for new directions in this domain, representing a beneficial endeavour in the development of platforms tailored for biological sequence classification. SBSM-Pro is available for access at http://lab.malab.cn/soft/SBSM-Pro/.
MechRetro is a chemical-mechanism-driven graph learning framework for interpretable retrosynthesis prediction and pathway planning
Oct 06, 2022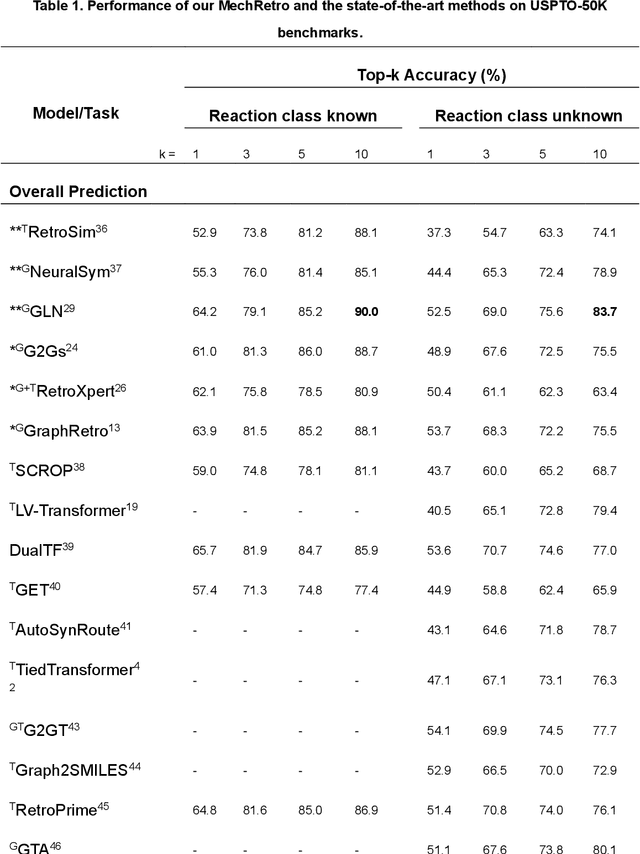
Leveraging artificial intelligence for automatic retrosynthesis speeds up organic pathway planning in digital laboratories. However, existing deep learning approaches are unexplainable, like "black box" with few insights, notably limiting their applications in real retrosynthesis scenarios. Here, we propose MechRetro, a chemical-mechanism-driven graph learning framework for interpretable retrosynthetic prediction and pathway planning, which learns several retrosynthetic actions to simulate a reverse reaction via elaborate self-adaptive joint learning. By integrating chemical knowledge as prior information, we design a novel Graph Transformer architecture to adaptively learn discriminative and chemically meaningful molecule representations, highlighting the strong capacity in molecule feature representation learning. We demonstrate that MechRetro outperforms the state-of-the-art approaches for retrosynthetic prediction with a large margin on large-scale benchmark datasets. Extending MechRetro to the multi-step retrosynthesis analysis, we identify efficient synthetic routes via an interpretable reasoning mechanism, leading to a better understanding in the realm of knowledgeable synthetic chemists. We also showcase that MechRetro discovers a novel pathway for protokylol, along with energy scores for uncertainty assessment, broadening the applicability for practical scenarios. Overall, we expect MechRetro to provide meaningful insights for high-throughput automated organic synthesis in drug discovery.
Reference-Based Sequence Classification
May 17, 2019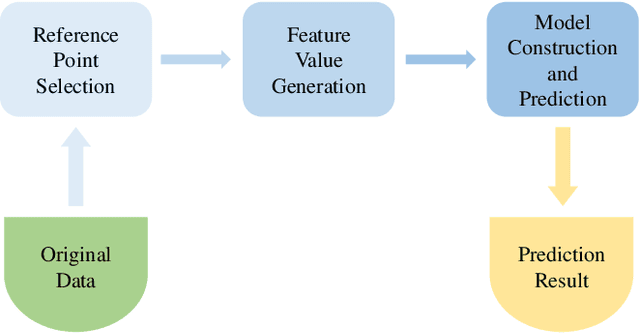
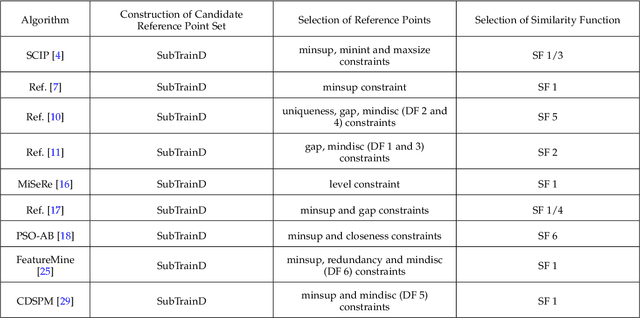
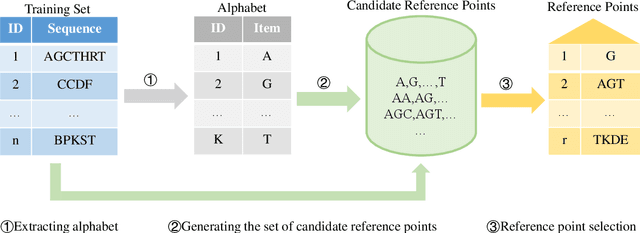
Sequence classification is an important data mining task in many real world applications. Over the past few decades, many sequence classification methods have been proposed from different aspects. In particular, the pattern-based method is one of the most important and widely studied sequence classification methods in the literature. In this paper, we present a reference-based sequence classification framework, which can unify existing pattern-based sequence classification methods under the same umbrella. More importantly, this framework can be used as a general platform for developing new sequence classification algorithms. By utilizing this framework as a tool, we propose new sequence classification algorithms that are quite different from existing solutions. Experimental results show that new methods developed under the proposed framework are capable of achieving comparable classification accuracy to those state-of-the-art sequence classification algorithms.
Instance-Based Classification through Hypothesis Testing
Jan 03, 2019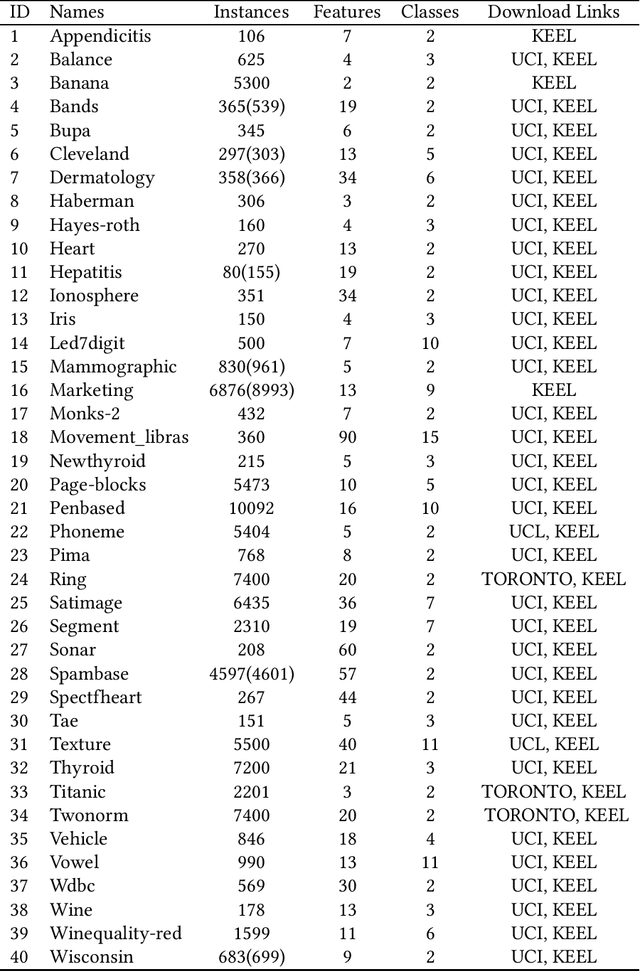
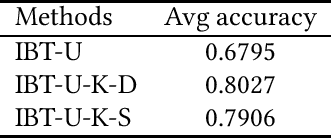

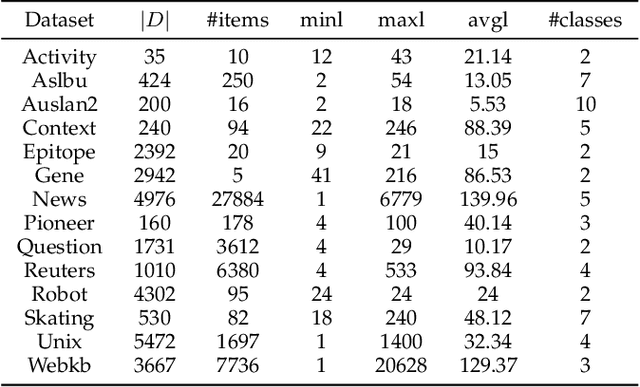
Classification is a fundamental problem in machine learning and data mining. During the past decades, numerous classification methods have been presented based on different principles. However, most existing classifiers cast the classification problem as an optimization problem and do not address the issue of statistical significance. In this paper, we formulate the binary classification problem as a two-sample testing problem. More precisely, our classification model is a generic framework that is composed of two steps. In the first step, the distance between the test instance and each training instance is calculated to derive two distance sets. In the second step, the two-sample test is performed under the null hypothesis that the two sets of distances are drawn from the same cumulative distribution. After these two steps, we have two p-values for each test instance and the test instance is assigned to the class associated with the smaller p-value. Essentially, the presented classification method can be regarded as an instance-based classifier based on hypothesis testing. The experimental results on 40 real data sets show that our method is able to achieve the same level performance as the state-of-the-art classifiers and has significantly better performance than existing testing-based classifiers. Furthermore, we can handle outlying instances and control the false discovery rate of test instances assigned to each class under the same framework.
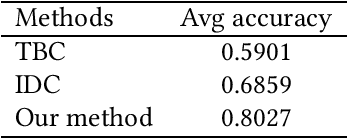
HPSLPred: An Ensemble Multi-label Classifier for Human Protein Subcellular Location Prediction with Imbalanced Source
Apr 18, 2017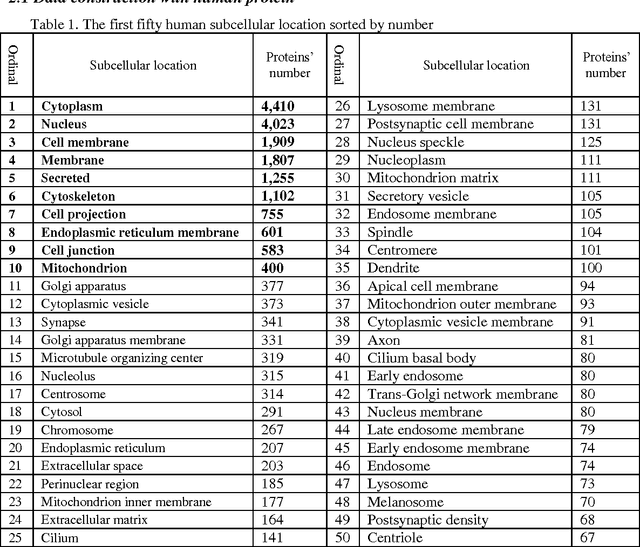
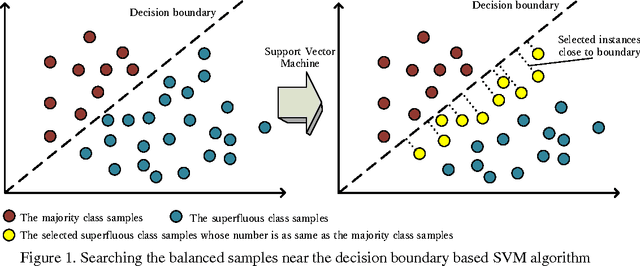
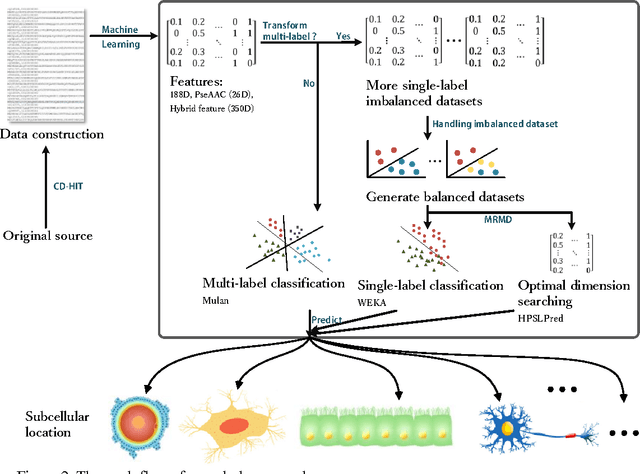
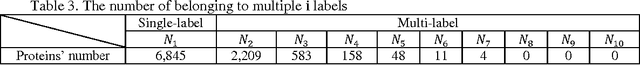
Predicting the subcellular localization of proteins is an important and challenging problem. Traditional experimental approaches are often expensive and time-consuming. Consequently, a growing number of research efforts employ a series of machine learning approaches to predict the subcellular location of proteins. There are two main challenges among the state-of-the-art prediction methods. First, most of the existing techniques are designed to deal with multi-class rather than multi-label classification, which ignores connections between multiple labels. In reality, multiple locations of particular proteins implies that there are vital and unique biological significances that deserve special focus and cannot be ignored. Second, techniques for handling imbalanced data in multi-label classification problems are necessary, but never employed. For solving these two issues, we have developed an ensemble multi-label classifier called HPSLPred, which can be applied for multi-label classification with an imbalanced protein source. For convenience, a user-friendly webserver has been established at http://server.malab.cn/HPSLPred.
Pretata: predicting TATA binding proteins with novel features and dimensionality reduction strategy
Mar 07, 2017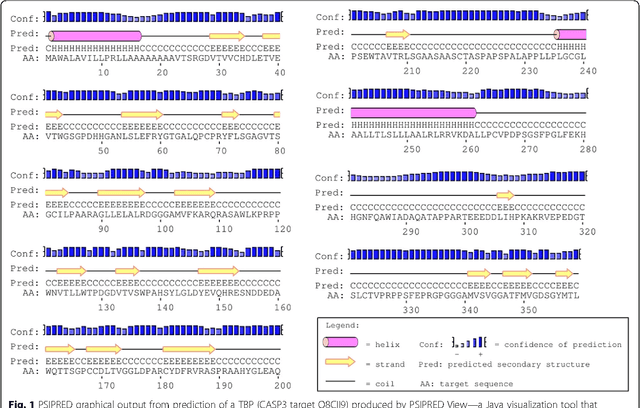
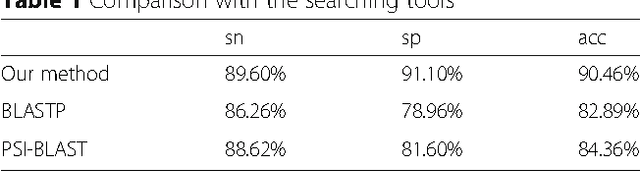
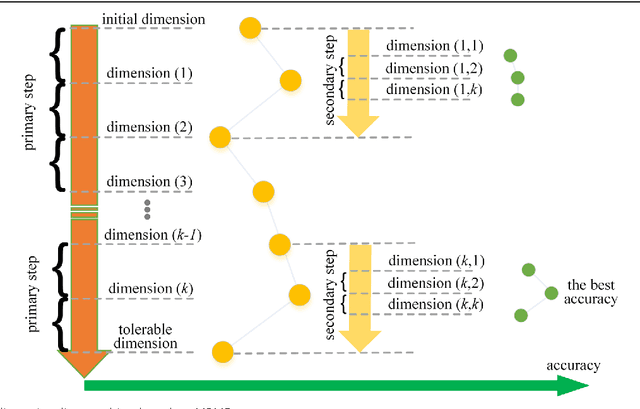
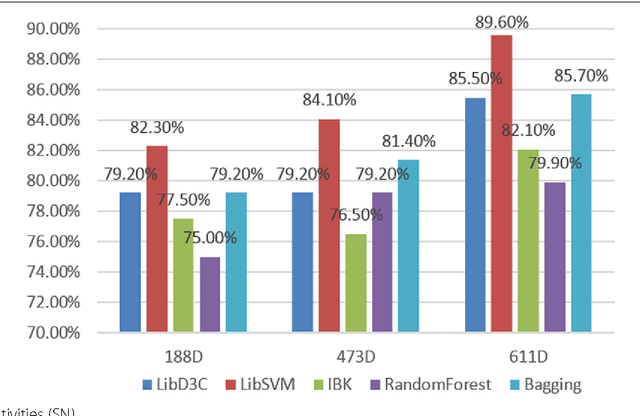
Background: It is necessary and essential to discovery protein function from the novel primary sequences. Wet lab experimental procedures are not only time-consuming, but also costly, so predicting protein structure and function reliably based only on amino acid sequence has significant value. TATA-binding protein (TBP) is a kind of DNA binding protein, which plays a key role in the transcription regulation. Our study proposed an automatic approach for identifying TATA-binding proteins efficiently, accurately, and conveniently. This method would guide for the special protein identification with computational intelligence strategies. Results: Firstly, we proposed novel fingerprint features for TBP based on pseudo amino acid composition, physicochemical properties, and secondary structure. Secondly, hierarchical features dimensionality reduction strategies were employed to improve the performance furthermore. Currently, Pretata achieves 92.92% TATA- binding protein prediction accuracy, which is better than all other existing methods. Conclusions: The experiments demonstrate that our method could greatly improve the prediction accuracy and speed, thus allowing large-scale NGS data prediction to be practical. A web server is developed to facilitate the other researchers, which can be accessed at http://server.malab.cn/preTata/.