 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMolCAP: Molecular Chemical reActivity pretraining and prompted-finetuning enhanced molecular representation learning
Jun 13, 2023



Molecular representation learning (MRL) is a fundamental task for drug discovery. However, previous deep-learning (DL) methods focus excessively on learning robust inner-molecular representations by mask-dominated pretraining framework, neglecting abundant chemical reactivity molecular relationships that have been demonstrated as the determining factor for various molecular property prediction tasks. Here, we present MolCAP to promote MRL, a graph pretraining Transformer based on chemical reactivity (IMR) knowledge with prompted finetuning. Results show that MolCAP outperforms comparative methods based on traditional molecular pretraining framework, in 13 publicly available molecular datasets across a diversity of biomedical tasks. Prompted by MolCAP, even basic graph neural networks are capable of achieving surprising performance that outperforms previous models, indicating the promising prospect of applying reactivity information for MRL. In addition, manual designed molecular templets are potential to uncover the dataset bias. All in all, we expect our MolCAP to gain more chemical meaningful insights for the entire process of drug discovery.
MechRetro is a chemical-mechanism-driven graph learning framework for interpretable retrosynthesis prediction and pathway planning
Oct 06, 2022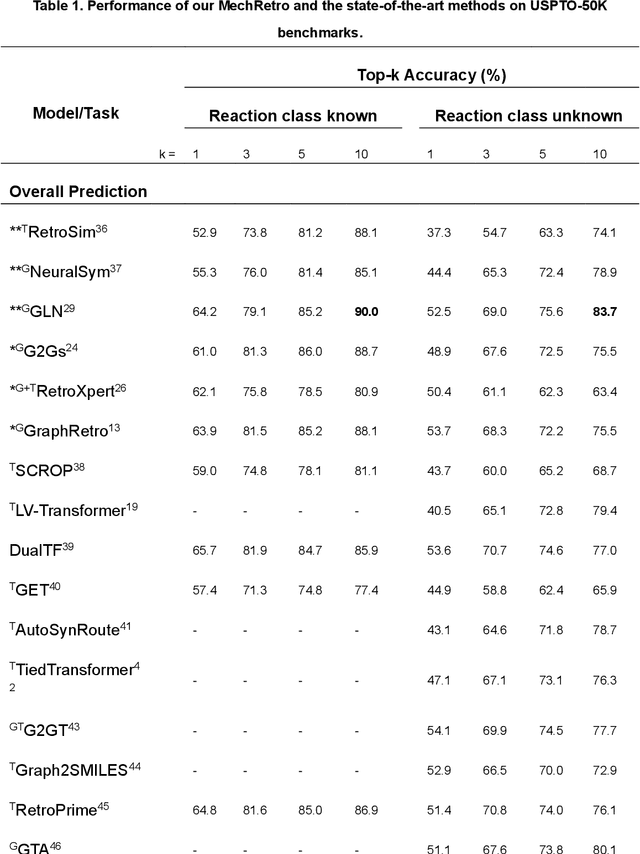
Leveraging artificial intelligence for automatic retrosynthesis speeds up organic pathway planning in digital laboratories. However, existing deep learning approaches are unexplainable, like "black box" with few insights, notably limiting their applications in real retrosynthesis scenarios. Here, we propose MechRetro, a chemical-mechanism-driven graph learning framework for interpretable retrosynthetic prediction and pathway planning, which learns several retrosynthetic actions to simulate a reverse reaction via elaborate self-adaptive joint learning. By integrating chemical knowledge as prior information, we design a novel Graph Transformer architecture to adaptively learn discriminative and chemically meaningful molecule representations, highlighting the strong capacity in molecule feature representation learning. We demonstrate that MechRetro outperforms the state-of-the-art approaches for retrosynthetic prediction with a large margin on large-scale benchmark datasets. Extending MechRetro to the multi-step retrosynthesis analysis, we identify efficient synthetic routes via an interpretable reasoning mechanism, leading to a better understanding in the realm of knowledgeable synthetic chemists. We also showcase that MechRetro discovers a novel pathway for protokylol, along with energy scores for uncertainty assessment, broadening the applicability for practical scenarios. Overall, we expect MechRetro to provide meaningful insights for high-throughput automated organic synthesis in drug discovery.