 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFocalOrder: Focal Preference Optimization for Reading Order Detection
Jan 12, 2026Reading order detection is the foundation of document understanding. Most existing methods rely on uniform supervision, implicitly assuming a constant difficulty distribution across layout regions. In this work, we challenge this assumption by revealing a critical flaw: \textbf{Positional Disparity}, a phenomenon where models demonstrate mastery over the deterministic start and end regions but suffer a performance collapse in the complex intermediate sections. This degradation arises because standard training allows the massive volume of easy patterns to drown out the learning signals from difficult layouts. To address this, we propose \textbf{FocalOrder}, a framework driven by \textbf{Focal Preference Optimization (FPO)}. Specifically, FocalOrder employs adaptive difficulty discovery with exponential moving average mechanism to dynamically pinpoint hard-to-learn transitions, while introducing a difficulty-calibrated pairwise ranking objective to enforce global logical consistency. Extensive experiments demonstrate that FocalOrder establishes new state-of-the-art results on OmniDocBench v1.0 and Comp-HRDoc. Our compact model not only outperforms competitive specialized baselines but also significantly surpasses large-scale general VLMs. These results demonstrate that aligning the optimization with intrinsic structural ambiguity of documents is critical for mastering complex document structures.
PARL: Position-Aware Relation Learning Network for Document Layout Analysis
Jan 12, 2026Document layout analysis aims to detect and categorize structural elements (e.g., titles, tables, figures) in scanned or digital documents. Popular methods often rely on high-quality Optical Character Recognition (OCR) to merge visual features with extracted text. This dependency introduces two major drawbacks: propagation of text recognition errors and substantial computational overhead, limiting the robustness and practical applicability of multimodal approaches. In contrast to the prevailing multimodal trend, we argue that effective layout analysis depends not on text-visual fusion, but on a deep understanding of documents' intrinsic visual structure. To this end, we propose PARL (Position-Aware Relation Learning Network), a novel OCR-free, vision-only framework that models layout through positional sensitivity and relational structure. Specifically, we first introduce a Bidirectional Spatial Position-Guided Deformable Attention module to embed explicit positional dependencies among layout elements directly into visual features. Second, we design a Graph Refinement Classifier (GRC) to refine predictions by modeling contextual relationships through a dynamically constructed layout graph. Extensive experiments show PARL achieves state-of-the-art results. It establishes a new benchmark for vision-only methods on DocLayNet and, notably, surpasses even strong multimodal models on M6Doc. Crucially, PARL (65M) is highly efficient, using roughly four times fewer parameters than large multimodal models (256M), demonstrating that sophisticated visual structure modeling can be both more efficient and robust than multimodal fusion.
GaussianEM: Model compositional and conformational heterogeneity using 3D Gaussians
Dec 25, 2025Understanding protein flexibility and its dynamic interactions with other molecules is essential for protein function study. Cryogenic electron microscopy (cryo-EM) provides an opportunity to directly observe macromolecular dynamics. However, analyzing datasets that contain both continuous motions and discrete states remains highly challenging. Here we present GaussianEM, a Gaussian pseudo-atomic framework that simultaneously models compositional and conformational heterogeneity from experimental cryo-EM images. GaussianEM employs a two-encoder-one-decoder architecture to map an image to its individual Gaussian components, and represent structural variability through changes in Gaussian parameters. This approach provides an intuitive and interpretable description of conformational changes, preserves local structural consistency along the transition trajectories, and naturally bridges the gap between density-based models and corresponding atomic models. We demonstrate the effectiveness of GaussianEM on both simulated and experimental datasets.
Simple but Effective Unsupervised Classification for Specified Domain Images: A Case Study on Fungi Images
Nov 15, 2023


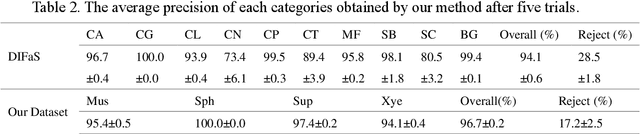
High-quality labeled datasets are essential for deep learning. Traditional manual annotation methods are not only costly and inefficient but also pose challenges in specialized domains where expert knowledge is needed. Self-supervised methods, despite leveraging unlabeled data for feature extraction, still require hundreds or thousands of labeled instances to guide the model for effective specialized image classification. Current unsupervised learning methods offer automatic classification without prior annotation but often compromise on accuracy. As a result, efficiently procuring high-quality labeled datasets remains a pressing challenge for specialized domain images devoid of annotated data. Addressing this, an unsupervised classification method with three key ideas is introduced: 1) dual-step feature dimensionality reduction using a pre-trained model and manifold learning, 2) a voting mechanism from multiple clustering algorithms, and 3) post-hoc instead of prior manual annotation. This approach outperforms supervised methods in classification accuracy, as demonstrated with fungal image data, achieving 94.1% and 96.7% on public and private datasets respectively. The proposed unsupervised classification method reduces dependency on pre-annotated datasets, enabling a closed-loop for data classification. The simplicity and ease of use of this method will also bring convenience to researchers in various fields in building datasets, promoting AI applications for images in specialized domains.
CryoAlign: feature-based method for global and local 3D alignment of EM density maps
Sep 17, 2023



Advances on cryo-electron imaging technologies have led to a rapidly increasing number of density maps. Alignment and comparison of density maps play a crucial role in interpreting structural information, such as conformational heterogeneity analysis using global alignment and atomic model assembly through local alignment. Here, we propose a fast and accurate global and local cryo-electron microscopy density map alignment method CryoAlign, which leverages local density feature descriptors to capture spatial structure similarities. CryoAlign is the first feature-based EM map alignment tool, in which the employment of feature-based architecture enables the rapid establishment of point pair correspondences and robust estimation of alignment parameters. Extensive experimental evaluations demonstrate the superiority of CryoAlign over the existing methods in both alignment accuracy and speed.
MFAI: A Scalable Bayesian Matrix Factorization Approach to Leveraging Auxiliary Information
Mar 05, 2023In various practical situations, matrix factorization methods suffer from poor data quality, such as high data sparsity and low signal-to-noise ratio (SNR). Here we consider a matrix factorization problem by utilizing auxiliary information, which is massively available in real applications, to overcome the challenges caused by poor data quality. Unlike existing methods that mainly rely on simple linear models to combine auxiliary information with the main data matrix, we propose to integrate gradient boosted trees in the probabilistic matrix factorization framework to effectively leverage auxiliary information (MFAI). Thus, MFAI naturally inherits several salient features of gradient boosted trees, such as the capability of flexibly modeling nonlinear relationships, and robustness to irrelevant features and missing values in auxiliary information. The parameters in MAFI can be automatically determined under the empirical Bayes framework, making it adaptive to the utilization of auxiliary information and immune to overfitting. Moreover, MFAI is computationally efficient and scalable to large-scale datasets by exploiting variational inference. We demonstrate the advantages of MFAI through comprehensive numerical results from simulation studies and real data analysis. Our approach is implemented in the R package mfair available at https://github.com/YangLabHKUST/mfair.
SHREC 2021: Classification in cryo-electron tomograms
Mar 18, 2022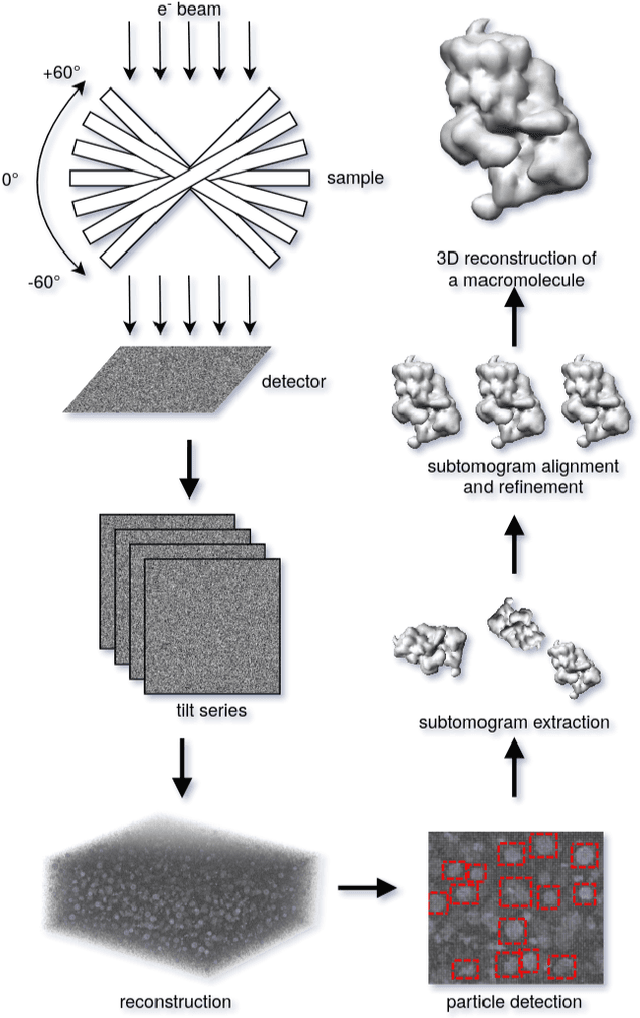
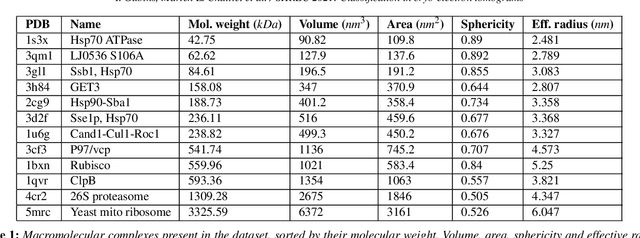
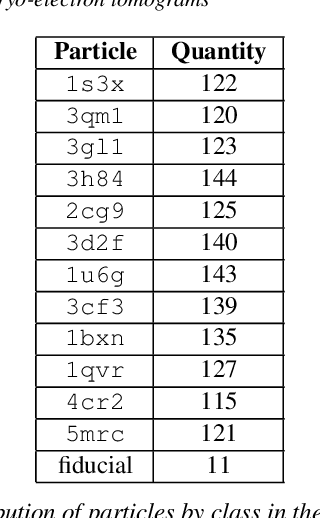
Cryo-electron tomography (cryo-ET) is an imaging technique that allows three-dimensional visualization of macro-molecular assemblies under near-native conditions. Cryo-ET comes with a number of challenges, mainly low signal-to-noise and inability to obtain images from all angles. Computational methods are key to analyze cryo-electron tomograms. To promote innovation in computational methods, we generate a novel simulated dataset to benchmark different methods of localization and classification of biological macromolecules in tomograms. Our publicly available dataset contains ten tomographic reconstructions of simulated cell-like volumes. Each volume contains twelve different types of complexes, varying in size, function and structure. In this paper, we have evaluated seven different methods of finding and classifying proteins. Seven research groups present results obtained with learning-based methods and trained on the simulated dataset, as well as a baseline template matching (TM), a traditional method widely used in cryo-ET research. We show that learning-based approaches can achieve notably better localization and classification performance than TM. We also experimentally confirm that there is a negative relationship between particle size and performance for all methods.
DWMD: Dimensional Weighted Orderwise Moment Discrepancy for Domain-specific Hidden Representation Matching
Jul 18, 2020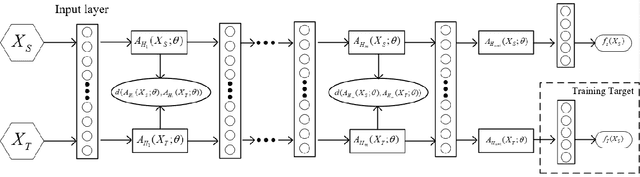
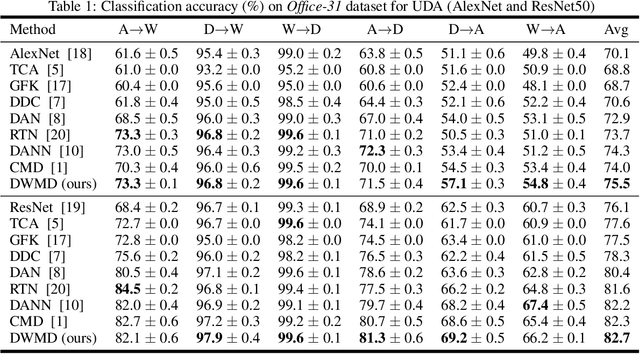
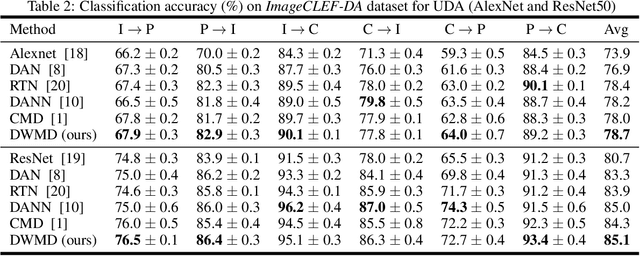
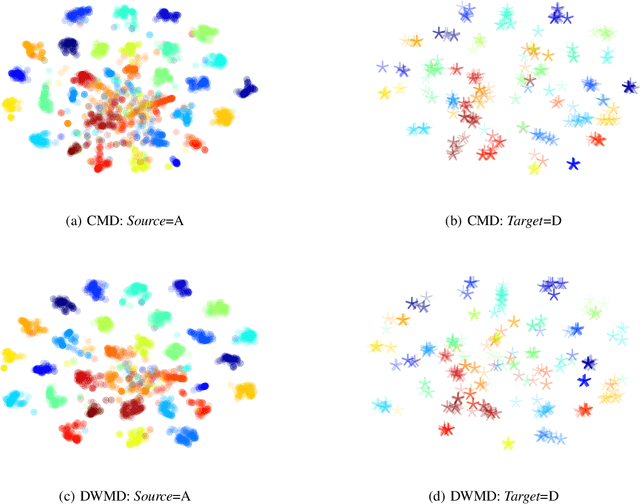
Knowledge transfer from a source domain to a different but semantically related target domain has long been an important topic in the context of unsupervised domain adaptation (UDA). A key challenge in this field is establishing a metric that can exactly measure the data distribution discrepancy between two homogeneous domains and adopt it in distribution alignment, especially in the matching of feature representations in the hidden activation space. Existing distribution matching approaches can be interpreted as failing to either explicitly orderwise align higher-order moments or satisfy the prerequisite of certain assumptions in practical uses. We propose a novel moment-based probability distribution metric termed dimensional weighted orderwise moment discrepancy (DWMD) for feature representation matching in the UDA scenario. Our metric function takes advantage of a series for high-order moment alignment, and we theoretically prove that our DWMD metric function is error-free, which means that it can strictly reflect the distribution differences between domains and is valid without any feature distribution assumption. In addition, since the discrepancies between probability distributions in each feature dimension are different, dimensional weighting is considered in our function. We further calculate the error bound of the empirical estimate of the DWMD metric in practical applications. Comprehensive experiments on benchmark datasets illustrate that our method yields state-of-the-art distribution metrics.
SegET: Deep Neural Network with Rich Contextual Features for Cellular Structures Segmentation in Electron Tomography Image
Nov 28, 2018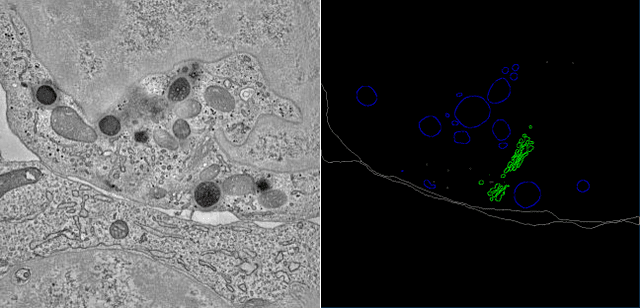
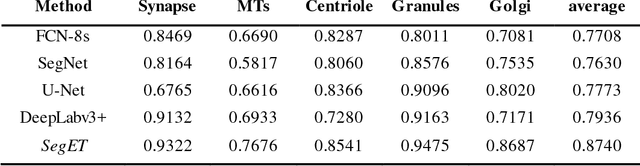
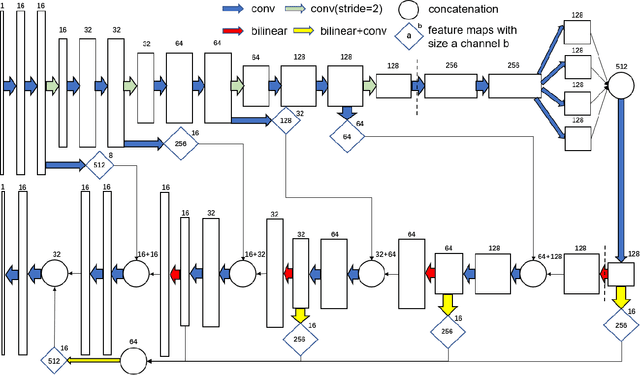
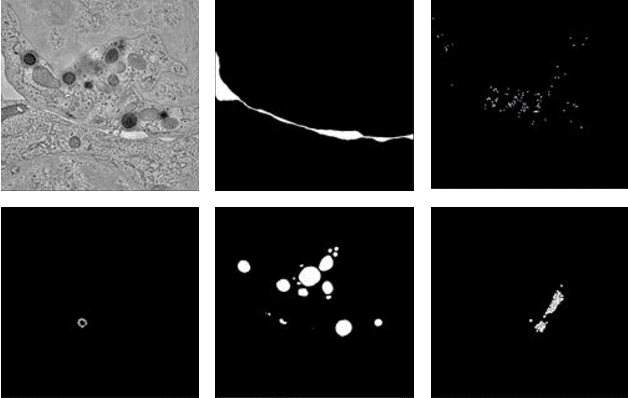
Electron tomography (ET) allows high-resolution reconstructions of macromolecular complexes at nearnative state. Cellular structures segmentation in the reconstruction data from electron tomographic images is often required for analyzing and visualizing biological structures, making it a powerful tool for quantitative descriptions of whole cell structures and understanding biological functions. However, these cellular structures are rather difficult to automatically separate or quantify from view owing to complex molecular environment and the limitations of reconstruction data of ET. In this paper, we propose a single end-to-end deep fully-convolutional semantic segmentation network dubbed SegET with rich contextual features which fully exploitsthe multi-scale and multi-level contextual information and reduces the loss of details of cellular structures in ET images. We trained and evaluated our network on the electron tomogram of the CTL Immunological Synapse from Cell Image library. Our results demonstrate that SegET can automatically segment accurately and outperform all other baseline methods on each individual structure in our ET dataset.
DLBI: Deep learning guided Bayesian inference for structure reconstruction of super-resolution fluorescence microscopy
Sep 01, 2018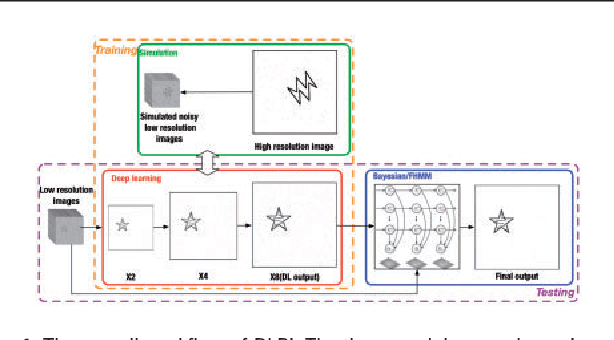
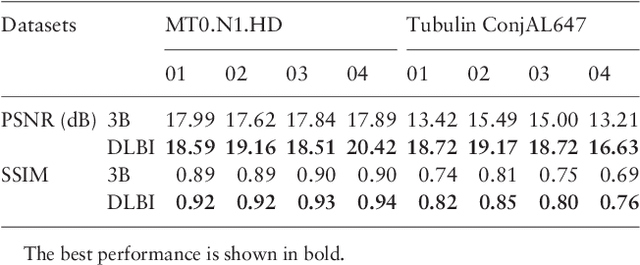
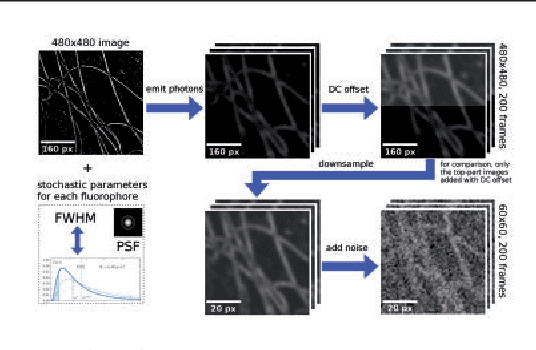
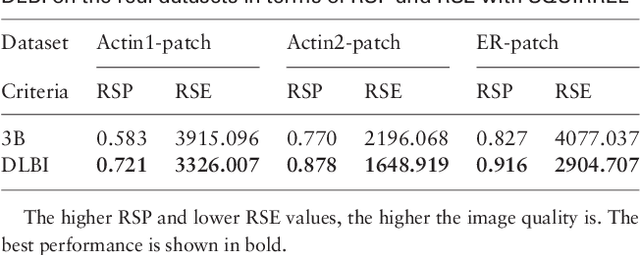
Super-resolution fluorescence microscopy, with a resolution beyond the diffraction limit of light, has become an indispensable tool to directly visualize biological structures in living cells at a nanometer-scale resolution. Despite advances in high-density super-resolution fluorescent techniques, existing methods still have bottlenecks, including extremely long execution time, artificial thinning and thickening of structures, and lack of ability to capture latent structures. Here we propose a novel deep learning guided Bayesian inference approach, DLBI, for the time-series analysis of high-density fluorescent images. Our method combines the strength of deep learning and statistical inference, where deep learning captures the underlying distribution of the fluorophores that are consistent with the observed time-series fluorescent images by exploring local features and correlation along time-axis, and statistical inference further refines the ultrastructure extracted by deep learning and endues physical meaning to the final image. Comprehensive experimental results on both real and simulated datasets demonstrate that our method provides more accurate and realistic local patch and large-field reconstruction than the state-of-the-art method, the 3B analysis, while our method is more than two orders of magnitude faster. The main program is available at https://github.com/lykaust15/DLBI
* Accepted by ISMB 2018