 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeState-of-the-art Small Language Coder Model: Mify-Coder
Dec 26, 2025We present Mify-Coder, a 2.5B-parameter code model trained on 4.2T tokens using a compute-optimal strategy built on the Mify-2.5B foundation model. Mify-Coder achieves comparable accuracy and safety while significantly outperforming much larger baseline models on standard coding and function-calling benchmarks, demonstrating that compact models can match frontier-grade models in code generation and agent-driven workflows. Our training pipeline combines high-quality curated sources with synthetic data generated through agentically designed prompts, refined iteratively using enterprise-grade evaluation datasets. LLM-based quality filtering further enhances data density, enabling frugal yet effective training. Through disciplined exploration of CPT-SFT objectives, data mixtures, and sampling dynamics, we deliver frontier-grade code intelligence within a single continuous training trajectory. Empirical evidence shows that principled data and compute discipline allow smaller models to achieve competitive accuracy, efficiency, and safety compliance. Quantized variants of Mify-Coder enable deployment on standard desktop environments without requiring specialized hardware.
ML-Dev-Bench: Comparative Analysis of AI Agents on ML development workflows
Feb 03, 2025



In this report, we present ML-Dev-Bench, a benchmark aimed at testing agentic capabilities on applied Machine Learning development tasks. While existing benchmarks focus on isolated coding tasks or Kaggle-style competitions, ML-Dev-Bench tests agents' ability to handle the full complexity of ML development workflows. The benchmark assesses performance across critical aspects including dataset handling, model training, improving existing models, debugging, and API integration with popular ML tools. We evaluate three agents -- ReAct, Openhands, and AIDE -- on a diverse set of 25 tasks, providing insights into their strengths and limitations in handling practical ML development challenges.
Interpretability analysis on a pathology foundation model reveals biologically relevant embeddings across modalities
Jul 15, 2024Mechanistic interpretability has been explored in detail for large language models (LLMs). For the first time, we provide a preliminary investigation with similar interpretability methods for medical imaging. Specifically, we analyze the features from a ViT-Small encoder obtained from a pathology Foundation Model via application to two datasets: one dataset of pathology images, and one dataset of pathology images paired with spatial transcriptomics. We discover an interpretable representation of cell and tissue morphology, along with gene expression within the model embedding space. Our work paves the way for further exploration around interpretable feature dimensions and their utility for medical and clinical applications.
PLUTO: Pathology-Universal Transformer
May 13, 2024



Pathology is the study of microscopic inspection of tissue, and a pathology diagnosis is often the medical gold standard to diagnose disease. Pathology images provide a unique challenge for computer-vision-based analysis: a single pathology Whole Slide Image (WSI) is gigapixel-sized and often contains hundreds of thousands to millions of objects of interest across multiple resolutions. In this work, we propose PathoLogy Universal TransfOrmer (PLUTO): a light-weight pathology FM that is pre-trained on a diverse dataset of 195 million image tiles collected from multiple sites and extracts meaningful representations across multiple WSI scales that enable a large variety of downstream pathology tasks. In particular, we design task-specific adaptation heads that utilize PLUTO's output embeddings for tasks which span pathology scales ranging from subcellular to slide-scale, including instance segmentation, tile classification, and slide-level prediction. We compare PLUTO's performance to other state-of-the-art methods on a diverse set of external and internal benchmarks covering multiple biologically relevant tasks, tissue types, resolutions, stains, and scanners. We find that PLUTO matches or outperforms existing task-specific baselines and pathology-specific foundation models, some of which use orders-of-magnitude larger datasets and model sizes when compared to PLUTO. Our findings present a path towards a universal embedding to power pathology image analysis, and motivate further exploration around pathology foundation models in terms of data diversity, architectural improvements, sample efficiency, and practical deployability in real-world applications.
ContriMix: Unsupervised disentanglement of content and attribute for domain generalization in microscopy image analysis
Jul 03, 2023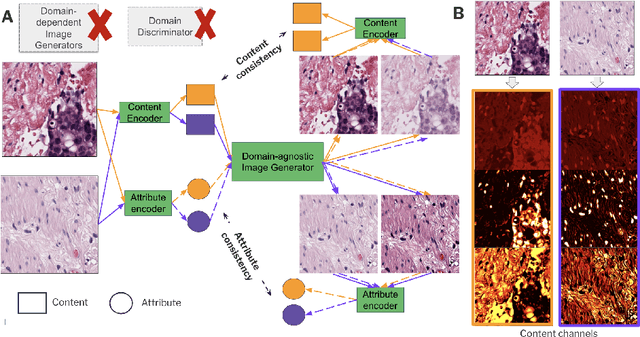


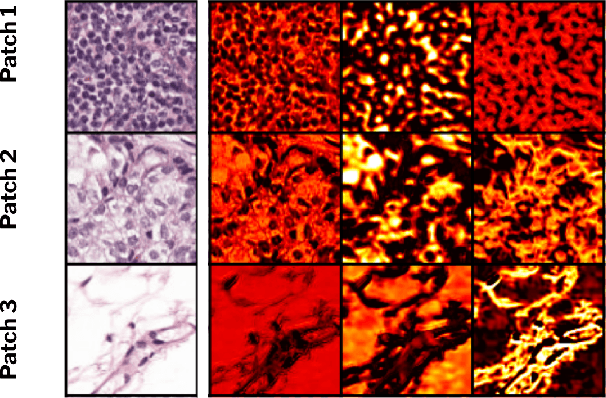
Domain generalization is critical for real-world applications of machine learning models to microscopy images, including histopathology and fluorescence imaging. Artifacts in histopathology arise through a complex combination of factors relating to tissue collection and laboratory processing, as well as factors intrinsic to patient samples. In fluorescence imaging, these artifacts stem from variations across experimental batches. The complexity and subtlety of these artifacts make the enumeration of data domains intractable. Therefore, augmentation-based methods of domain generalization that require domain identifiers and manual fine-tuning are inadequate in this setting. To overcome this challenge, we introduce ContriMix, a domain generalization technique that learns to generate synthetic images by disentangling and permuting the biological content ("content") and technical variations ("attributes") in microscopy images. ContriMix does not rely on domain identifiers or handcrafted augmentations and makes no assumptions about the input characteristics of images. We assess the performance of ContriMix on two pathology datasets (Camelyon17-WILDS and a prostate cell classification dataset) and one fluorescence microscopy dataset (RxRx1-WILDS). ContriMix outperforms current state-of-the-art methods in all datasets, motivating its usage for microscopy image analysis in real-world settings where domain information is hard to come by.
Synthetic DOmain-Targeted Augmentation (S-DOTA) Improves Model Generalization in Digital Pathology
May 03, 2023Machine learning algorithms have the potential to improve patient outcomes in digital pathology. However, generalization of these tools is currently limited by sensitivity to variations in tissue preparation, staining procedures and scanning equipment that lead to domain shift in digitized slides. To overcome this limitation and improve model generalization, we studied the effectiveness of two Synthetic DOmain-Targeted Augmentation (S-DOTA) methods, namely CycleGAN-enabled Scanner Transform (ST) and targeted Stain Vector Augmentation (SVA), and compared them against the International Color Consortium (ICC) profile-based color calibration (ICC Cal) method and a baseline method using traditional brightness, color and noise augmentations. We evaluated the ability of these techniques to improve model generalization to various tasks and settings: four models, two model types (tissue segmentation and cell classification), two loss functions, six labs, six scanners, and three indications (hepatocellular carcinoma (HCC), nonalcoholic steatohepatitis (NASH), prostate adenocarcinoma). We compared these methods based on the macro-averaged F1 scores on in-distribution (ID) and out-of-distribution (OOD) test sets across multiple domains, and found that S-DOTA methods (i.e., ST and SVA) led to significant improvements over ICC Cal and baseline on OOD data while maintaining comparable performance on ID data. Thus, we demonstrate that S-DOTA may help address generalization due to domain shift in real world applications.
SC-MIL: Supervised Contrastive Multiple Instance Learning for Imbalanced Classification in Pathology
Mar 23, 2023Multiple Instance learning (MIL) models have been extensively used in pathology to predict biomarkers and risk-stratify patients from gigapixel-sized images. Machine learning problems in medical imaging often deal with rare diseases, making it important for these models to work in a label-imbalanced setting. Furthermore, these imbalances can occur in out-of-distribution (OOD) datasets when the models are deployed in the real-world. We leverage the idea that decoupling feature and classifier learning can lead to improved decision boundaries for label imbalanced datasets. To this end, we investigate the integration of supervised contrastive learning with multiple instance learning (SC-MIL). Specifically, we propose a joint-training MIL framework in the presence of label imbalance that progressively transitions from learning bag-level representations to optimal classifier learning. We perform experiments with different imbalance settings for two well-studied problems in cancer pathology: subtyping of non-small cell lung cancer and subtyping of renal cell carcinoma. SC-MIL provides large and consistent improvements over other techniques on both in-distribution (ID) and OOD held-out sets across multiple imbalanced settings.
Self-training of Machine Learning Models for Liver Histopathology: Generalization under Clinical Shifts
Nov 14, 2022


Histopathology images are gigapixel-sized and include features and information at different resolutions. Collecting annotations in histopathology requires highly specialized pathologists, making it expensive and time-consuming. Self-training can alleviate annotation constraints by learning from both labeled and unlabeled data, reducing the amount of annotations required from pathologists. We study the design of teacher-student self-training systems for Non-alcoholic Steatohepatitis (NASH) using clinical histopathology datasets with limited annotations. We evaluate the models on in-distribution and out-of-distribution test data under clinical data shifts. We demonstrate that through self-training, the best student model statistically outperforms the teacher with a $3\%$ absolute difference on the macro F1 score. The best student model also approaches the performance of a fully supervised model trained with twice as many annotations.
Finding Patient Zero: Learning Contagion Source with Graph Neural Networks
Jun 27, 2020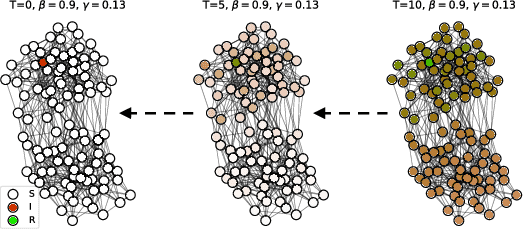
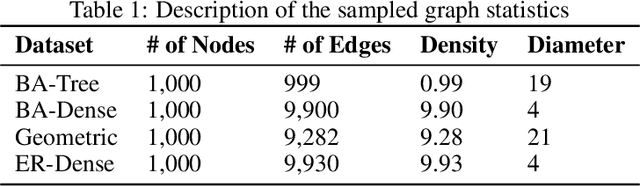
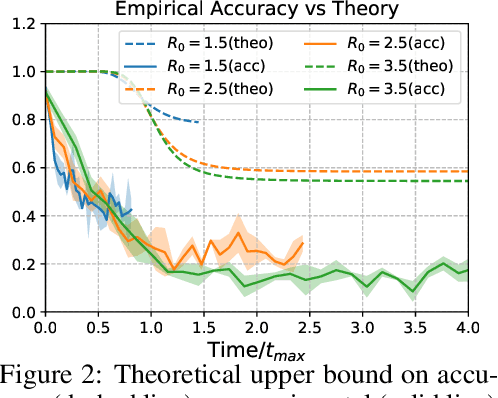
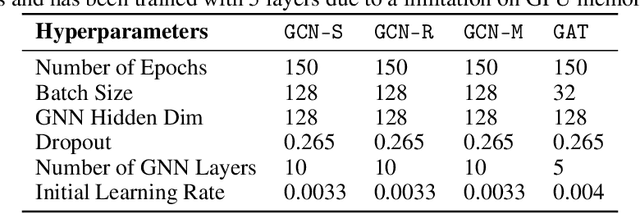
Locating the source of an epidemic, or patient zero (P0), can provide critical insights into the infection's transmission course and allow efficient resource allocation. Existing methods use graph-theoretic centrality measures and expensive message-passing algorithms, requiring knowledge of the underlying dynamics and its parameters. In this paper, we revisit this problem using graph neural networks (GNNs) to learn P0. We establish a theoretical limit for the identification of P0 in a class of epidemic models. We evaluate our method against different epidemic models on both synthetic and a real-world contact network considering a disease with history and characteristics of COVID-19. % We observe that GNNs can identify P0 close to the theoretical bound on accuracy, without explicit input of dynamics or its parameters. In addition, GNN is over 100 times faster than classic methods for inference on arbitrary graph topologies. Our theoretical bound also shows that the epidemic is like a ticking clock, emphasizing the importance of early contact-tracing. We find a maximum time after which accurate recovery of the source becomes impossible, regardless of the algorithm used.