 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHidden Citations Obscure True Impact in Science
Oct 24, 2023References, the mechanism scientists rely on to signal previous knowledge, lately have turned into widely used and misused measures of scientific impact. Yet, when a discovery becomes common knowledge, citations suffer from obliteration by incorporation. This leads to the concept of hidden citation, representing a clear textual credit to a discovery without a reference to the publication embodying it. Here, we rely on unsupervised interpretable machine learning applied to the full text of each paper to systematically identify hidden citations. We find that for influential discoveries hidden citations outnumber citation counts, emerging regardless of publishing venue and discipline. We show that the prevalence of hidden citations is not driven by citation counts, but rather by the degree of the discourse on the topic within the text of the manuscripts, indicating that the more discussed is a discovery, the less visible it is to standard bibliometric analysis. Hidden citations indicate that bibliometric measures offer a limited perspective on quantifying the true impact of a discovery, raising the need to extract knowledge from the full text of the scientific corpus.
AI-Bind: Improving Binding Predictions for Novel Protein Targets and Ligands
Dec 28, 2021
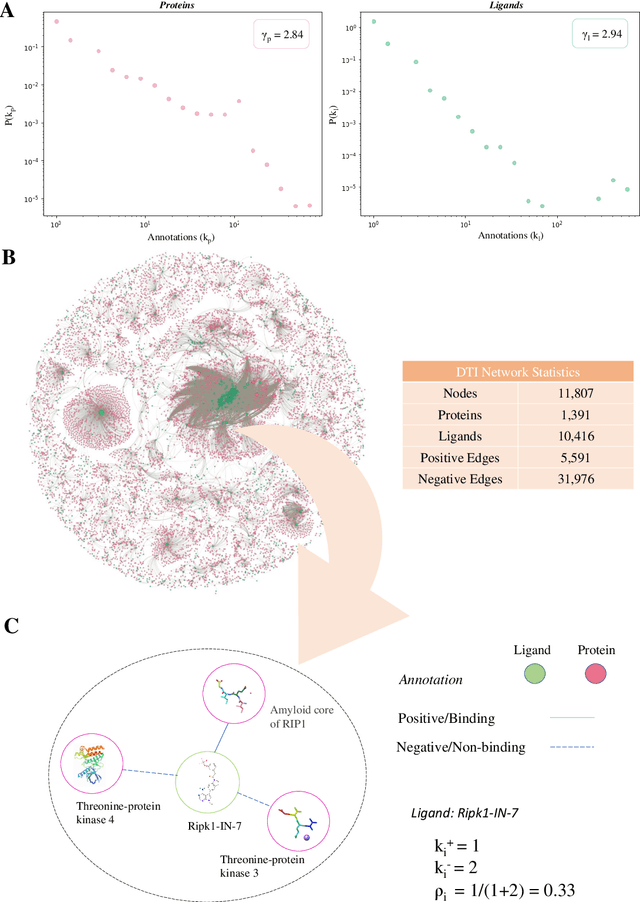

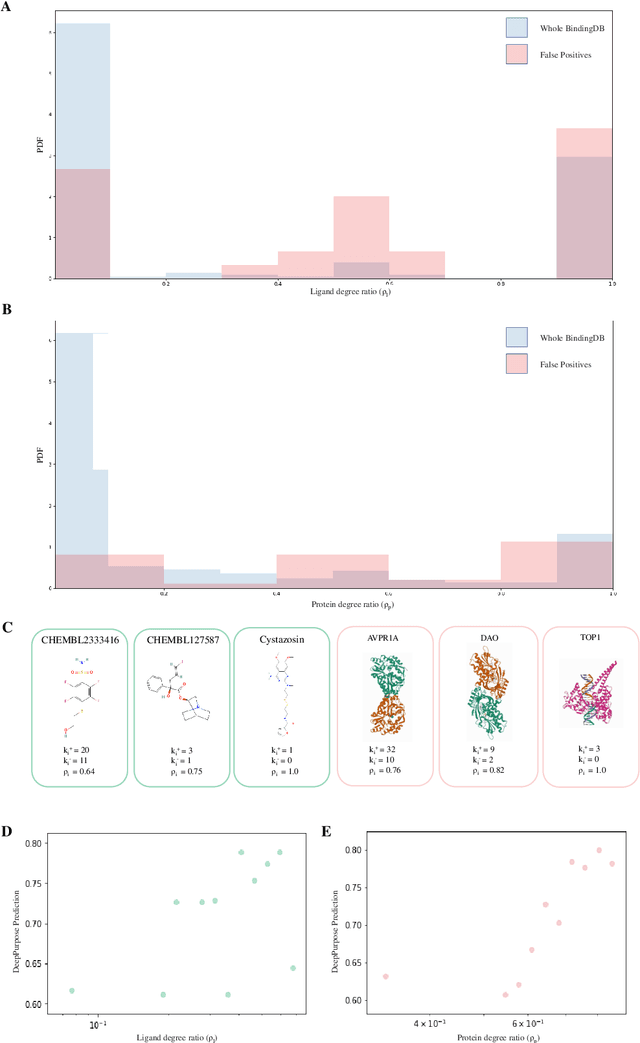
Identifying novel drug-target interactions (DTI) is a critical and rate limiting step in drug discovery. While deep learning models have been proposed to accelerate the identification process, we show that state-of-the-art models fail to generalize to novel (i.e., never-before-seen) structures. We first unveil the mechanisms responsible for this shortcoming, demonstrating how models rely on shortcuts that leverage the topology of the protein-ligand bipartite network, rather than learning the node features. Then, we introduce AI-Bind, a pipeline that combines network-based sampling strategies with unsupervised pre-training, allowing us to limit the annotation imbalance and improve binding predictions for novel proteins and ligands. We illustrate the value of AI-Bind by predicting drugs and natural compounds with binding affinity to SARS-CoV-2 viral proteins and the associated human proteins. We also validate these predictions via auto-docking simulations and comparison with recent experimental evidence. Overall, AI-Bind offers a powerful high-throughput approach to identify drug-target combinations, with the potential of becoming a powerful tool in drug discovery.
3D Topology Transformation with Generative Adversarial Networks
Jul 07, 2020
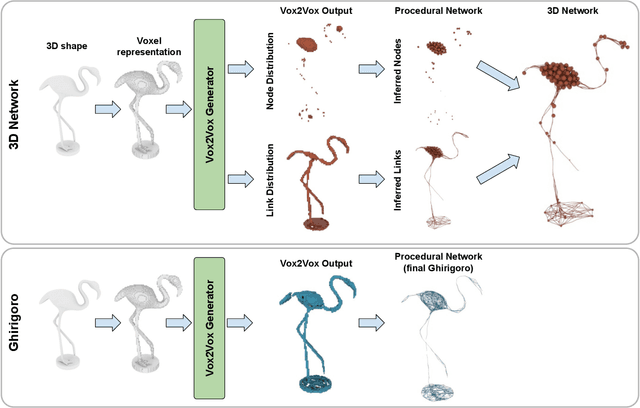
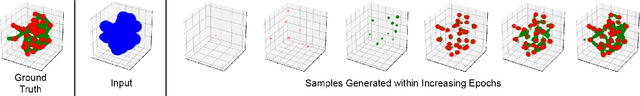
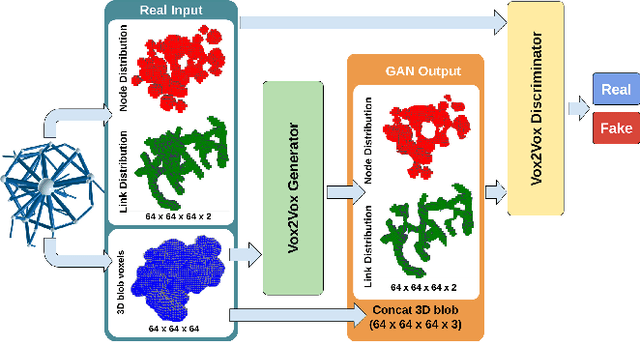
Generation and transformation of images and videos using artificial intelligence have flourished over the past few years. Yet, there are only a few works aiming to produce creative 3D shapes, such as sculptures. Here we show a novel 3D-to-3D topology transformation method using Generative Adversarial Networks (GAN). We use a modified pix2pix GAN, which we call Vox2Vox, to transform the volumetric style of a 3D object while retaining the original object shape. In particular, we show how to transform 3D models into two new volumetric topologies - the 3D Network and the Ghirigoro. We describe how to use our approach to construct customized 3D representations. We believe that the generated 3D shapes are novel and inspirational. Finally, we compare the results between our approach and a baseline algorithm that directly convert the 3D shapes, without using our GAN.
Finding Patient Zero: Learning Contagion Source with Graph Neural Networks
Jun 27, 2020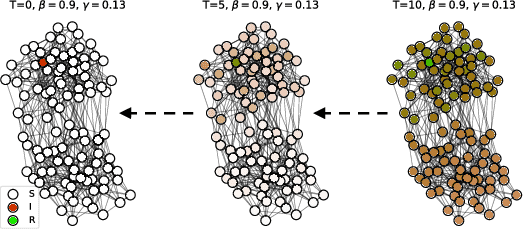
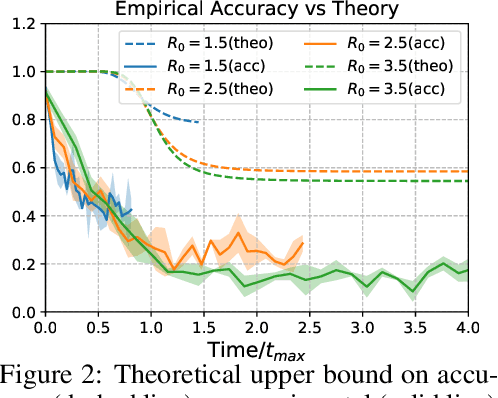
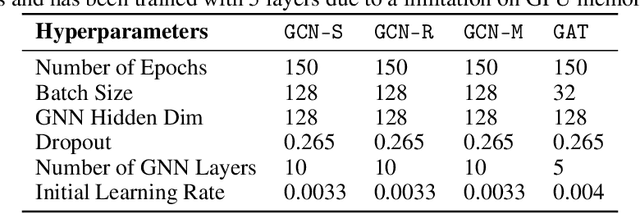
Locating the source of an epidemic, or patient zero (P0), can provide critical insights into the infection's transmission course and allow efficient resource allocation. Existing methods use graph-theoretic centrality measures and expensive message-passing algorithms, requiring knowledge of the underlying dynamics and its parameters. In this paper, we revisit this problem using graph neural networks (GNNs) to learn P0. We establish a theoretical limit for the identification of P0 in a class of epidemic models. We evaluate our method against different epidemic models on both synthetic and a real-world contact network considering a disease with history and characteristics of COVID-19. % We observe that GNNs can identify P0 close to the theoretical bound on accuracy, without explicit input of dynamics or its parameters. In addition, GNN is over 100 times faster than classic methods for inference on arbitrary graph topologies. Our theoretical bound also shows that the epidemic is like a ticking clock, emphasizing the importance of early contact-tracing. We find a maximum time after which accurate recovery of the source becomes impossible, regardless of the algorithm used.
Network Medicine Framework for Identifying Drug Repurposing Opportunities for COVID-19
Apr 15, 2020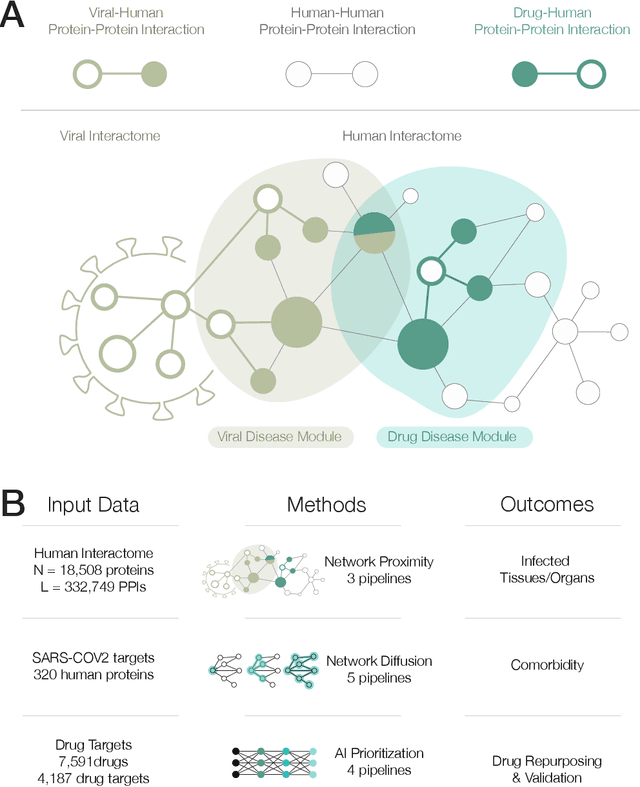
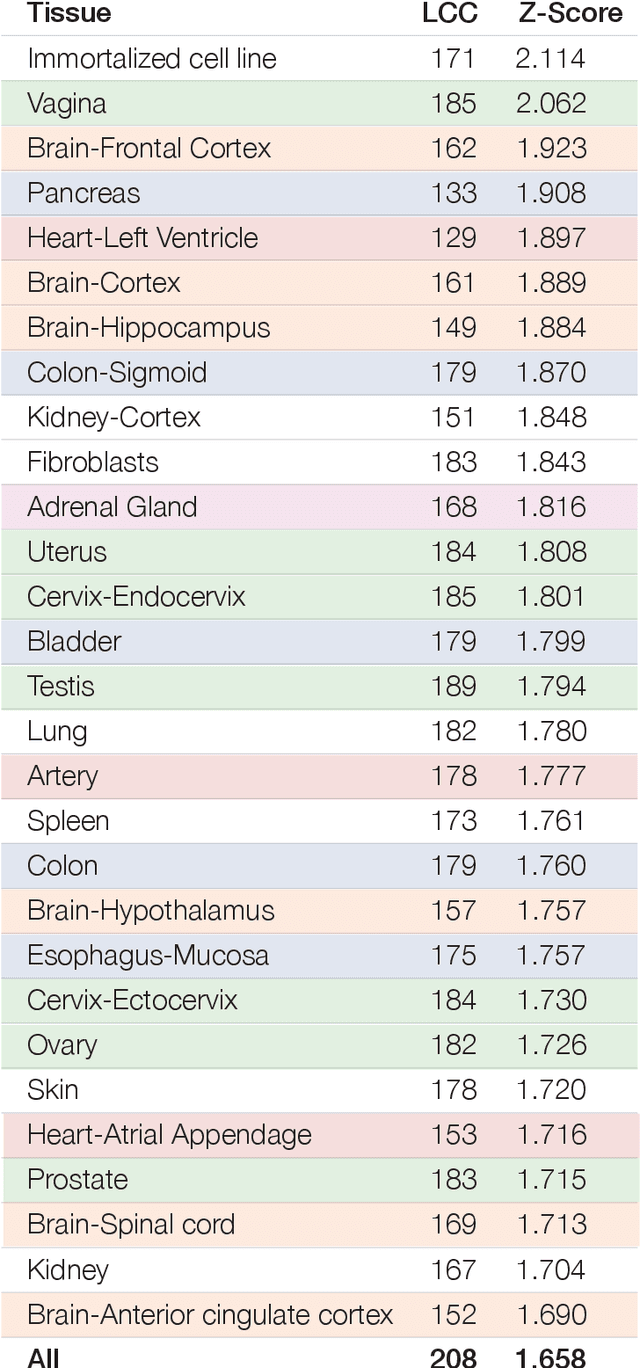
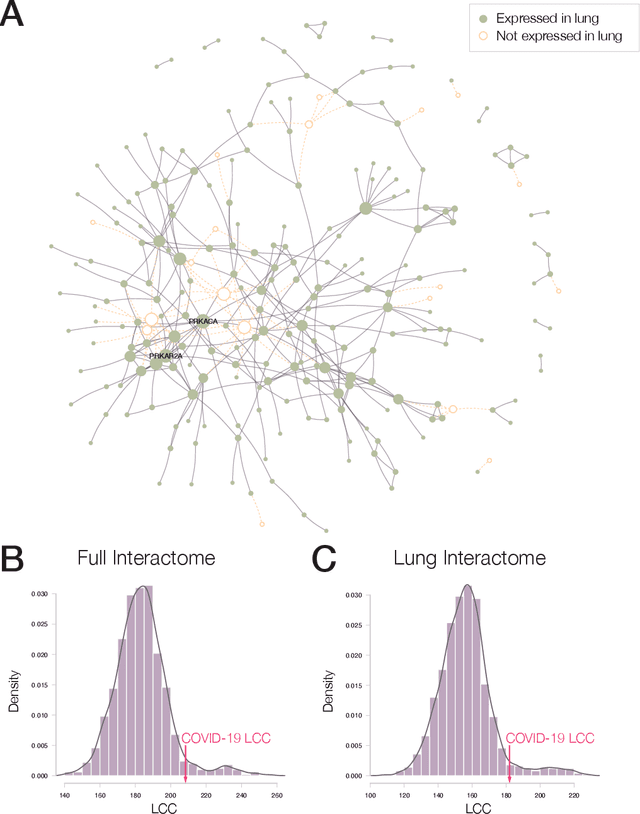
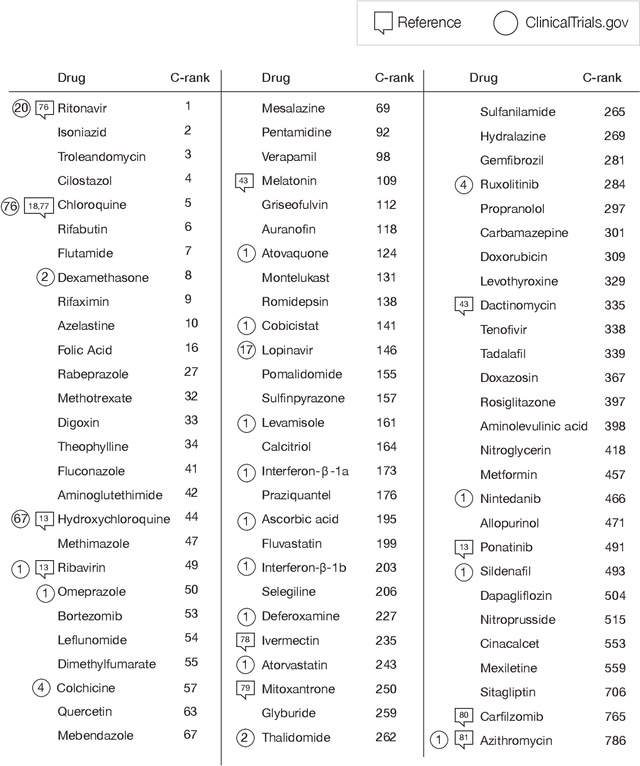
The COVID-19 pandemic demands the rapid identification of drug-repurpusing candidates. In the past decade, network medicine had developed a framework consisting of a series of quantitative approaches and predictive tools to study host-pathogen interactions, unveil the molecular mechanisms of the infection, identify comorbidities as well as rapidly detect drug repurpusing candidates. Here, we adapt the network-based toolset to COVID-19, recovering the primary pulmonary manifestations of the virus in the lung as well as observed comorbidities associated with cardiovascular diseases. We predict that the virus can manifest itself in other tissues, such as the reproductive system, and brain regions, moreover we predict neurological comorbidities. We build on these findings to deploy three network-based drug repurposing strategies, relying on network proximity, diffusion, and AI-based metrics, allowing to rank all approved drugs based on their likely efficacy for COVID-19 patients, aggregate all predictions, and, thereby to arrive at 81 promising repurposing candidates. We validate the accuracy of our predictions using drugs currently in clinical trials, and an expression-based validation of selected candidates suggests that these drugs, with known toxicities and side effects, could be moved to clinical trials rapidly.
Understanding the Representation Power of Graph Neural Networks in Learning Graph Topology
Jul 11, 2019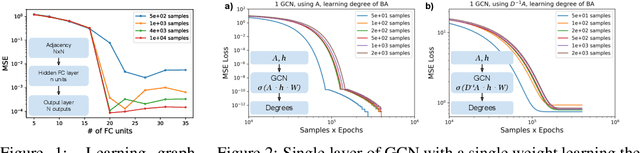

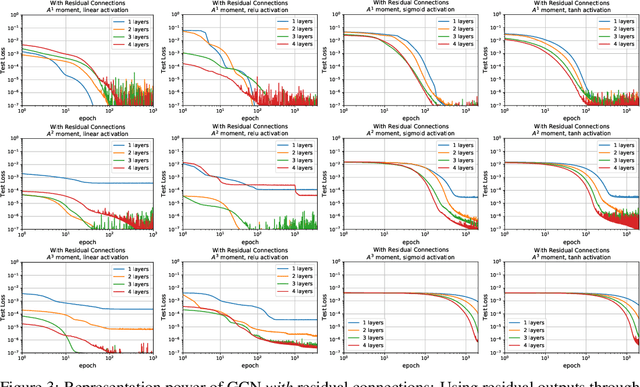
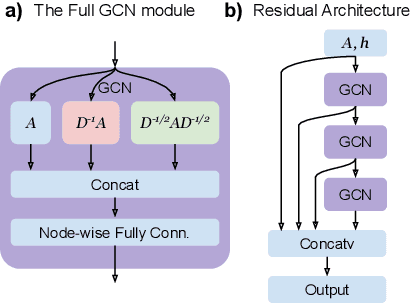
To deepen our understanding of graph neural networks, we investigate the representation power of Graph Convolutional Networks (GCN) through the looking glass of graph moments, a key property of graph topology encoding path of various lengths. We find that GCNs are rather restrictive in learning graph moments. Without careful design, GCNs can fail miserably even with multiple layers and nonlinear activation functions. We analyze theoretically the expressiveness of GCNs, arriving at a modular GCN design, using different propagation rules. Our modular design is capable of distinguishing graphs from different graph generation models for surprisingly small graphs, a notoriously difficult problem in network science. Our investigation suggests that, depth is much more influential than width, with deeper GCNs being more capable of learning higher order graph moments. Additionally, combining GCN modules with different propagation rules is critical to the representation power of GCNs.