 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Large-Scale Dataset and Benchmark: Do Protein-Ligand Models Learn Binding Sites or Just Binding Likelihood?
May 21, 2026Protein-ligand modeling underpins computational drug discovery and molecular design. Existing protein-ligand benchmarks typically evaluate whether a protein and ligand interact and how strongly they bind, through tasks such as binary binding prediction and affinity regression. However, these evaluations provide limited evidence of whether models can localize binding sites or identify the non-covalent interactions underlying molecular recognition. To address this gap, we introduce InteractBind, a large-scale protein-ligand dataset comprising approximately 100k protein-ligand pairs, together with a benchmark for fine-grained evaluation. The core fine-grained task is that of binding-site localization, which uses protein-residue and ligand-atom interaction maps spanning six major types of non-covalent interactions to assess whether model-derived interaction maps localize binding sites. InteractBind further includes binding affinity and protein similarity-controlled splits to support realistic generalization assessment. Using InteractBind, we evaluate eight existing sequence-based and interaction-aware models, assessing binary binding prediction and binding-site localization. Results reveal limited binding-site localization despite strong binary binding prediction, with marked variation across non-covalent interaction types. Overall, InteractBind establishes a benchmark paradigm that encourages the development of more interpretable and physically grounded protein-ligand models.
One Patient, Many Contexts: Scaling Medical AI Through Contextual Intelligence
Jun 11, 2025

Medical foundation models, including language models trained on clinical notes, vision-language models on medical images, and multimodal models on electronic health records, can summarize clinical notes, answer medical questions, and assist in decision-making. Adapting these models to new populations, specialties, or settings typically requires fine-tuning, careful prompting, or retrieval from knowledge bases. This can be impractical, and limits their ability to interpret unfamiliar inputs and adjust to clinical situations not represented during training. As a result, models are prone to contextual errors, where predictions appear reasonable but fail to account for critical patient-specific or contextual information. These errors stem from a fundamental limitation that current models struggle with: dynamically adjusting their behavior across evolving contexts of medical care. In this Perspective, we outline a vision for context-switching in medical AI: models that dynamically adapt their reasoning without retraining to new specialties, populations, workflows, and clinical roles. We envision context-switching AI to diagnose, manage, and treat a wide range of diseases across specialties and regions, and expand access to medical care.
Disease Gene Prioritization With Quantum Walks
Nov 09, 2023

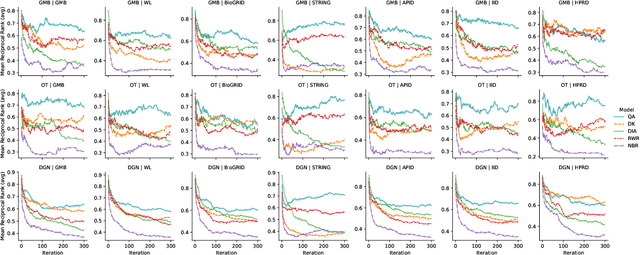

Disease gene prioritization assigns scores to genes or proteins according to their likely relevance for a given disease based on a provided set of seed genes. Here, we describe a new algorithm for disease gene prioritization based on continuous-time quantum walks using the adjacency matrix of a protein-protein interaction (PPI) network. Our algorithm can be seen as a quantum version of a previous method known as the diffusion kernel, but, importantly, has higher performance in predicting disease genes, and also permits the encoding of seed node self-loops into the underlying Hamiltonian, which offers yet another boost in performance. We demonstrate the success of our proposed method by comparing it to several well-known gene prioritization methods on three disease sets, across seven different PPI networks. In order to compare these methods, we use cross-validation and examine the mean reciprocal ranks and recall values. We further validate our method by performing an enrichment analysis of the predicted genes for coronary artery disease. We also investigate the impact of adding self-loops to the seeds, and argue that they allow the quantum walker to remain more local to low-degree seed nodes.
Network Medicine Framework for Identifying Drug Repurposing Opportunities for COVID-19
Apr 15, 2020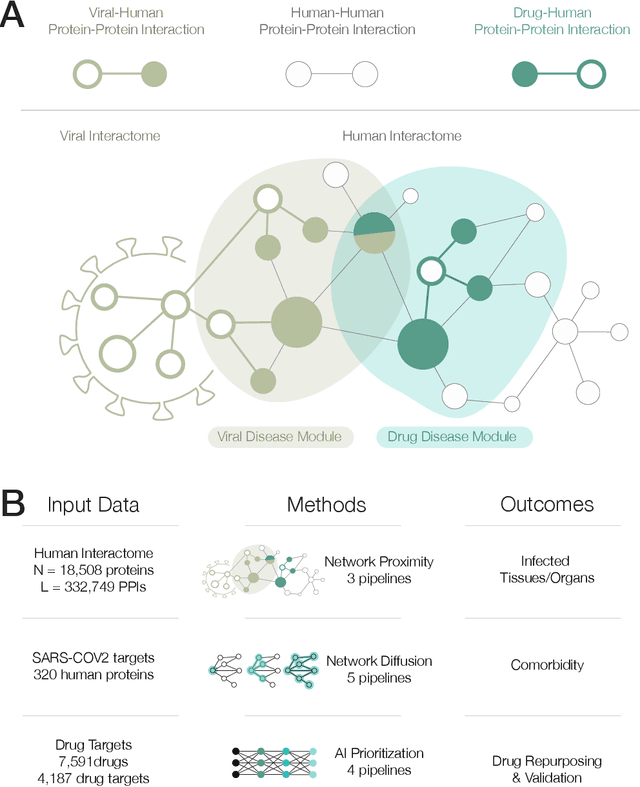
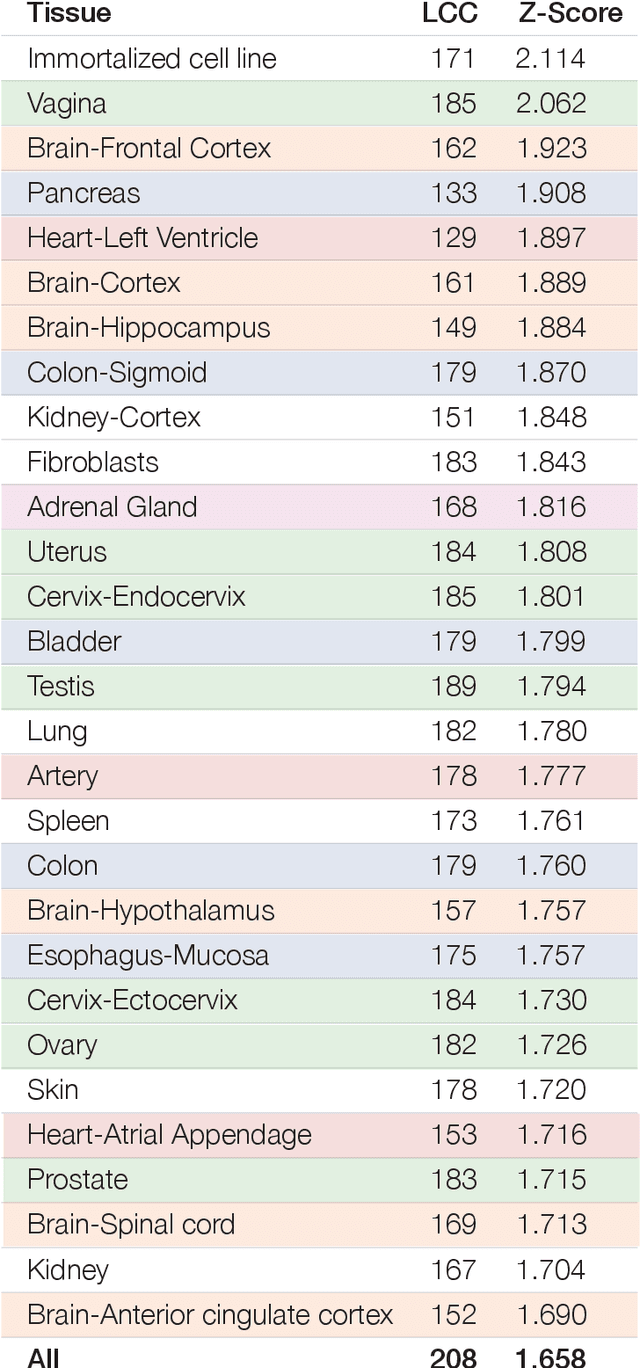
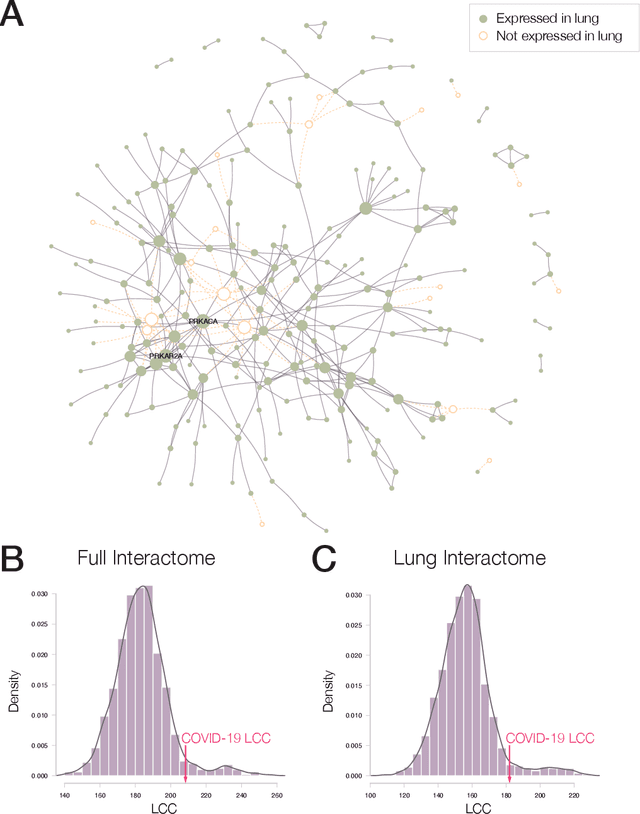
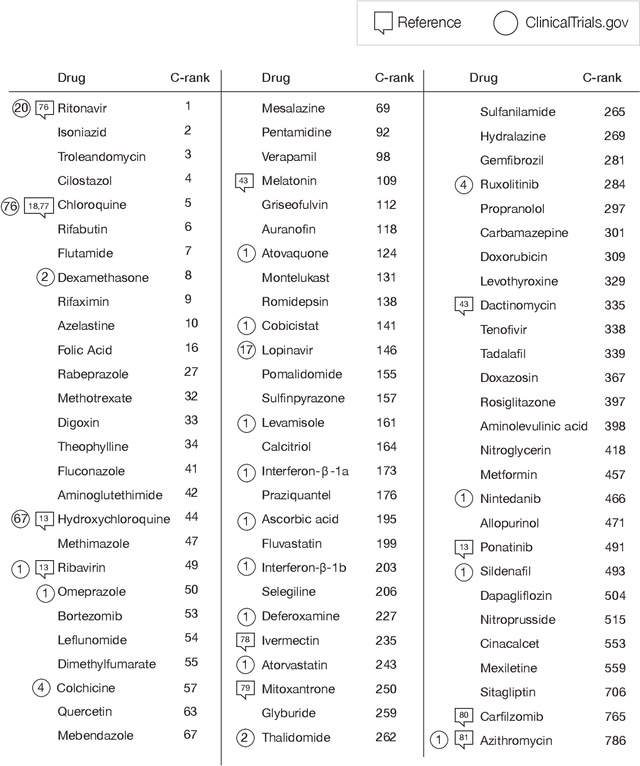
The COVID-19 pandemic demands the rapid identification of drug-repurpusing candidates. In the past decade, network medicine had developed a framework consisting of a series of quantitative approaches and predictive tools to study host-pathogen interactions, unveil the molecular mechanisms of the infection, identify comorbidities as well as rapidly detect drug repurpusing candidates. Here, we adapt the network-based toolset to COVID-19, recovering the primary pulmonary manifestations of the virus in the lung as well as observed comorbidities associated with cardiovascular diseases. We predict that the virus can manifest itself in other tissues, such as the reproductive system, and brain regions, moreover we predict neurological comorbidities. We build on these findings to deploy three network-based drug repurposing strategies, relying on network proximity, diffusion, and AI-based metrics, allowing to rank all approved drugs based on their likely efficacy for COVID-19 patients, aggregate all predictions, and, thereby to arrive at 81 promising repurposing candidates. We validate the accuracy of our predictions using drugs currently in clinical trials, and an expression-based validation of selected candidates suggests that these drugs, with known toxicities and side effects, could be moved to clinical trials rapidly.