 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph AI generates neurological hypotheses validated in molecular, organoid, and clinical systems
Dec 13, 2025Neurological diseases are the leading global cause of disability, yet most lack disease-modifying treatments. We present PROTON, a heterogeneous graph transformer that generates testable hypotheses across molecular, organoid, and clinical systems. To evaluate PROTON, we apply it to Parkinson's disease (PD), bipolar disorder (BD), and Alzheimer's disease (AD). In PD, PROTON linked genetic risk loci to genes essential for dopaminergic neuron survival and predicted pesticides toxic to patient-derived neurons, including the insecticide endosulfan, which ranked within the top 1.29% of predictions. In silico screens performed by PROTON reproduced six genome-wide $α$-synuclein experiments, including a split-ubiquitin yeast two-hybrid system (normalized enrichment score [NES] = 2.30, FDR-adjusted $p < 1 \times 10^{-4}$), an ascorbate peroxidase proximity labeling assay (NES = 2.16, FDR $< 1 \times 10^{-4}$), and a high-depth targeted exome sequencing study in 496 synucleinopathy patients (NES = 2.13, FDR $< 1 \times 10^{-4}$). In BD, PROTON predicted calcitriol as a candidate drug that reversed proteomic alterations observed in cortical organoids derived from BD patients. In AD, we evaluated PROTON predictions in health records from $n = 610,524$ patients at Mass General Brigham, confirming that five PROTON-predicted drugs were associated with reduced seven-year dementia risk (minimum hazard ratio = 0.63, 95% CI: 0.53-0.75, $p < 1 \times 10^{-7}$). PROTON generated neurological hypotheses that were evaluated across molecular, organoid, and clinical systems, defining a path for AI-driven discovery in neurological disease.
One Patient, Many Contexts: Scaling Medical AI Through Contextual Intelligence
Jun 11, 2025

Medical foundation models, including language models trained on clinical notes, vision-language models on medical images, and multimodal models on electronic health records, can summarize clinical notes, answer medical questions, and assist in decision-making. Adapting these models to new populations, specialties, or settings typically requires fine-tuning, careful prompting, or retrieval from knowledge bases. This can be impractical, and limits their ability to interpret unfamiliar inputs and adjust to clinical situations not represented during training. As a result, models are prone to contextual errors, where predictions appear reasonable but fail to account for critical patient-specific or contextual information. These errors stem from a fundamental limitation that current models struggle with: dynamically adjusting their behavior across evolving contexts of medical care. In this Perspective, we outline a vision for context-switching in medical AI: models that dynamically adapt their reasoning without retraining to new specialties, populations, workflows, and clinical roles. We envision context-switching AI to diagnose, manage, and treat a wide range of diseases across specialties and regions, and expand access to medical care.
Controllable Sequence Editing for Counterfactual Generation
Feb 05, 2025



Sequence models generate counterfactuals by modifying parts of a sequence based on a given condition, enabling reasoning about "what if" scenarios. While these models excel at conditional generation, they lack fine-grained control over when and where edits occur. Existing approaches either focus on univariate sequences or assume that interventions affect the entire sequence globally. However, many applications require precise, localized modifications, where interventions take effect only after a specified time and impact only a subset of co-occurring variables. We introduce CLEF, a controllable sequence editing model for counterfactual reasoning about both immediate and delayed effects. CLEF learns temporal concepts that encode how and when interventions should influence a sequence. With these concepts, CLEF selectively edits relevant time steps while preserving unaffected portions of the sequence. We evaluate CLEF on cellular and patient trajectory datasets, where gene regulation affects only certain genes at specific time steps, or medical interventions alter only a subset of lab measurements. CLEF improves immediate sequence editing by up to 36.01% in MAE compared to baselines. Unlike prior methods, CLEF enables one-step generation of counterfactual sequences at any future time step, outperforming baselines by up to 65.71% in MAE. A case study on patients with type 1 diabetes mellitus shows that CLEF identifies clinical interventions that shift patient trajectories toward healthier outcomes.
Graph AI in Medicine
Oct 20, 2023In clinical artificial intelligence (AI), graph representation learning, mainly through graph neural networks (GNNs), stands out for its capability to capture intricate relationships within structured clinical datasets. With diverse data -- from patient records to imaging -- GNNs process data holistically by viewing modalities as nodes interconnected by their relationships. Graph AI facilitates model transfer across clinical tasks, enabling models to generalize across patient populations without additional parameters or minimal re-training. However, the importance of human-centered design and model interpretability in clinical decision-making cannot be overstated. Since graph AI models capture information through localized neural transformations defined on graph relationships, they offer both an opportunity and a challenge in elucidating model rationale. Knowledge graphs can enhance interpretability by aligning model-driven insights with medical knowledge. Emerging graph models integrate diverse data modalities through pre-training, facilitate interactive feedback loops, and foster human-AI collaboration, paving the way to clinically meaningful predictions.
Discrepancies in Epidemiological Modeling of Aggregated Heterogeneous Data
Jun 20, 2021
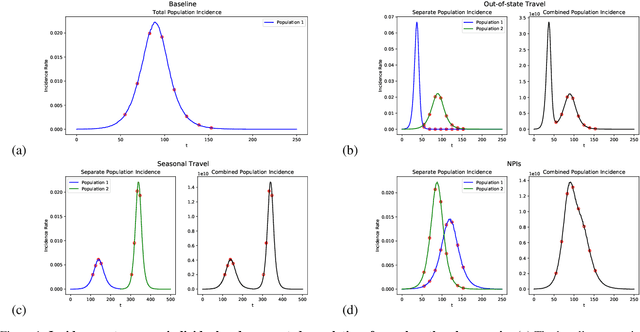

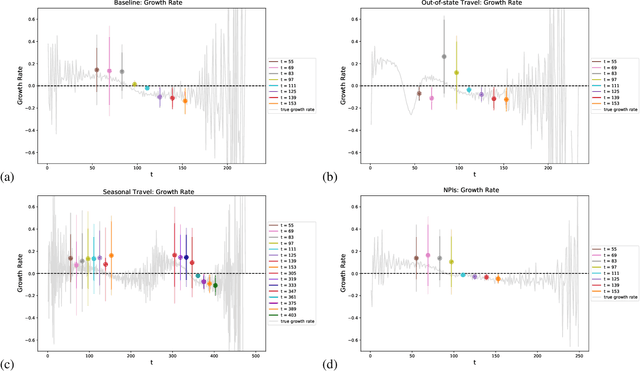
Within epidemiological modeling, the majority of analyses assume a single epidemic process for generating ground-truth data. However, this assumed data generation process can be unrealistic, since data sources for epidemics are often aggregated across geographic regions and communities. As a result, state-of-the-art models for estimating epidemiological parameters, e.g.~transmission rates, can be inappropriate when faced with complex systems. Our work empirically demonstrates some limitations of applying epidemiological models to aggregated datasets. We generate three complex outbreak scenarios by combining incidence curves from multiple epidemics that are independently simulated via SEIR models with different sets of parameters. Using these scenarios, we assess the robustness of a state-of-the-art Bayesian inference method that estimates the epidemic trajectory from viral load surveillance data. We evaluate two data-generating models within this Bayesian inference framework: a simple exponential growth model and a highly flexible Gaussian process prior model. Our results show that both models generate accurate transmission rate estimates for the combined incidence curve at the cost of generating biased estimates for each underlying epidemic, reflecting highly heterogeneous underlying population dynamics. The exponential growth model, while interpretable, is unable to capture the complexity of the underlying epidemics. With sufficient surveillance data, the Gaussian process prior model captures the shape of complex trajectories, but is imprecise for periods of low data coverage. Thus, our results highlight the potential pitfalls of neglecting complexity and heterogeneity in the data generation process, which can mask underlying location- and population-specific epidemic dynamics.
Deep Contextual Learners for Protein Networks
Jun 04, 2021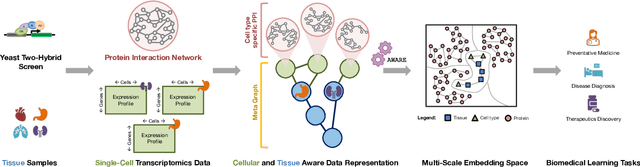

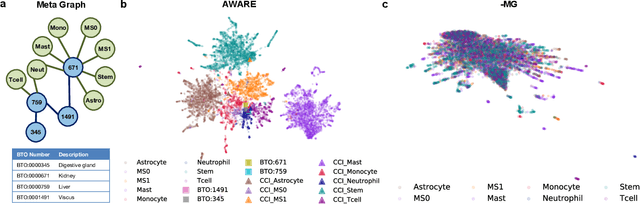
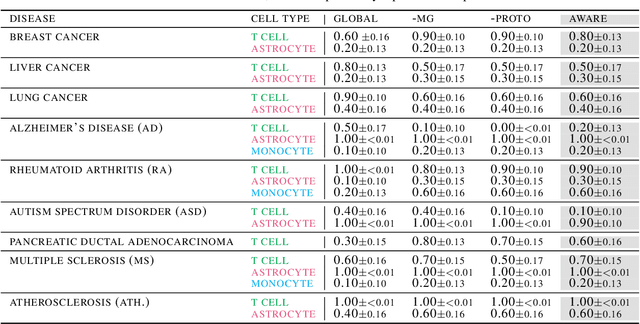
Spatial context is central to understanding health and disease. Yet reference protein interaction networks lack such contextualization, thereby limiting the study of where protein interactions likely occur in the human body. Contextualized protein interactions could better characterize genes with disease-specific interactions and elucidate diseases' manifestation in specific cell types. Here, we introduce AWARE, a graph neural message passing approach to inject cellular and tissue context into protein embeddings. AWARE optimizes for a multi-scale embedding space, whose structure reflects the topology of cell type specific networks. We construct a multi-scale network of the Human Cell Atlas and apply AWARE to learn protein, cell type, and tissue embeddings that uphold cell type and tissue hierarchies. We demonstrate AWARE on the novel task of predicting whether a gene is associated with a disease and where it most likely manifests in the human body. AWARE embeddings outperform global embeddings by at least 12.5%, highlighting the importance of contextual learners for protein networks.
Representation Learning for Networks in Biology and Medicine: Advancements, Challenges, and Opportunities
Apr 11, 2021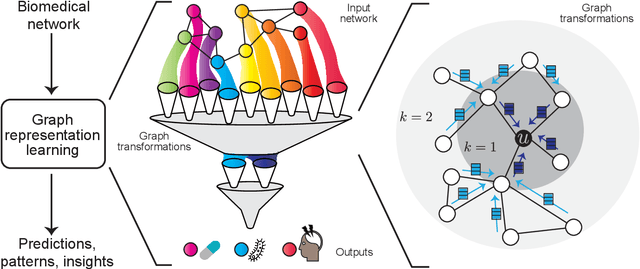
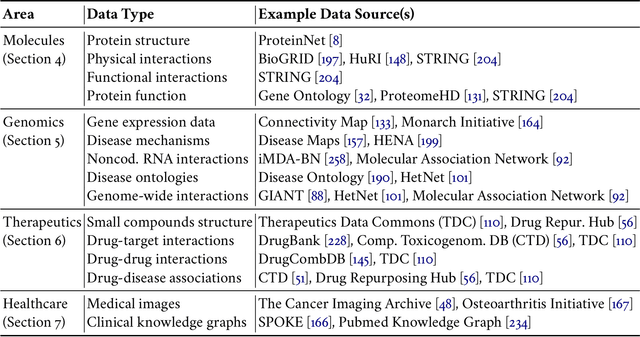
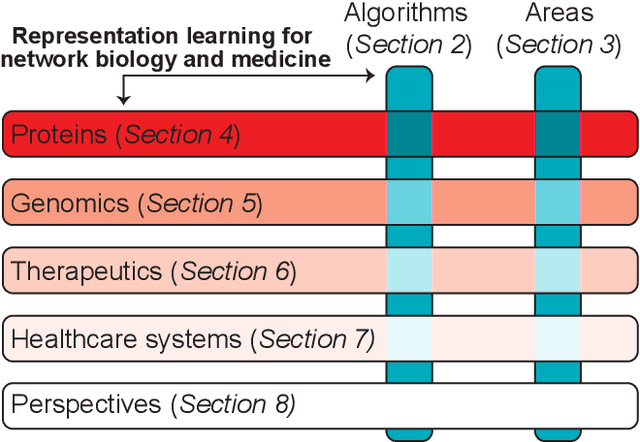
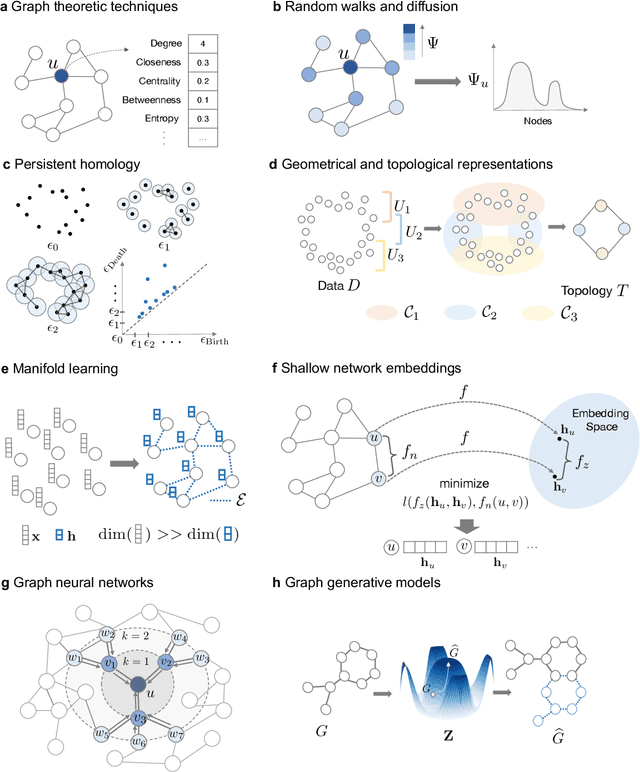
With the remarkable success of representation learning in providing powerful predictions and data insights, we have witnessed a rapid expansion of representation learning techniques into modeling, analysis, and learning with networks. Biomedical networks are universal descriptors of systems of interacting elements, from protein interactions to disease networks, all the way to healthcare systems and scientific knowledge. In this review, we put forward an observation that long-standing principles of network biology and medicine -- while often unspoken in machine learning research -- can provide the conceptual grounding for representation learning, explain its current successes and limitations, and inform future advances. We synthesize a spectrum of algorithmic approaches that, at their core, leverage topological features to embed networks into compact vector spaces. We also provide a taxonomy of biomedical areas that are likely to benefit most from algorithmic innovation. Representation learning techniques are becoming essential for identifying causal variants underlying complex traits, disentangling behaviors of single cells and their impact on health, and diagnosing and treating diseases with safe and effective medicines.
Subgraph Neural Networks
Jun 19, 2020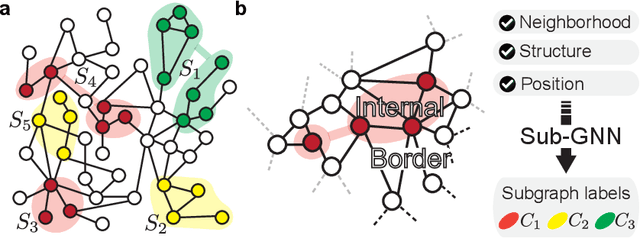

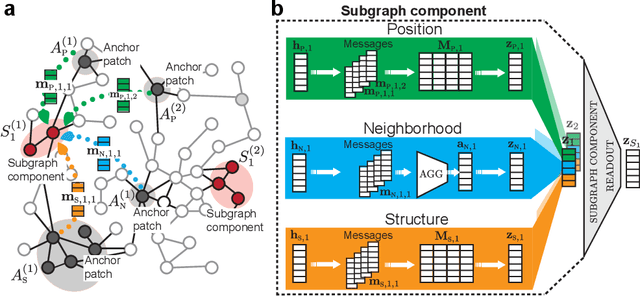
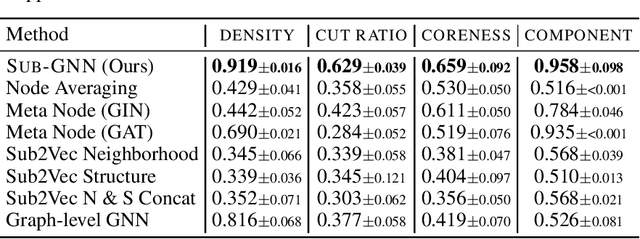
Deep learning methods for graphs achieve remarkable performance on many node-level and graph-level prediction tasks. However, despite the proliferation of the methods and their success, prevailing Graph Neural Networks (GNNs) neglect subgraphs, rendering subgraph prediction tasks challenging to tackle in many impactful applications. Further, subgraph prediction tasks present several unique challenges, because subgraphs can have non-trivial internal topology, but also carry a notion of position and external connectivity information relative to the underlying graph in which they exist. Here, we introduce SUB-GNN, a subgraph neural network to learn disentangled subgraph representations. In particular, we propose a novel subgraph routing mechanism that propagates neural messages between the subgraph's components and randomly sampled anchor patches from the underlying graph, yielding highly accurate subgraph representations. SUB-GNN specifies three channels, each designed to capture a distinct aspect of subgraph structure, and we provide empirical evidence that the channels encode their intended properties. We design a series of new synthetic and real-world subgraph datasets. Empirical results for subgraph classification on eight datasets show that SUB-GNN achieves considerable performance gains, outperforming strong baseline methods, including node-level and graph-level GNNs, by 12.4% over the strongest baseline. SUB-GNN performs exceptionally well on challenging biomedical datasets when subgraphs have complex topology and even comprise multiple disconnected components.