 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeuralPLexer3: Accurate Biomolecular Complex Structure Prediction with Flow Models
Dec 18, 2024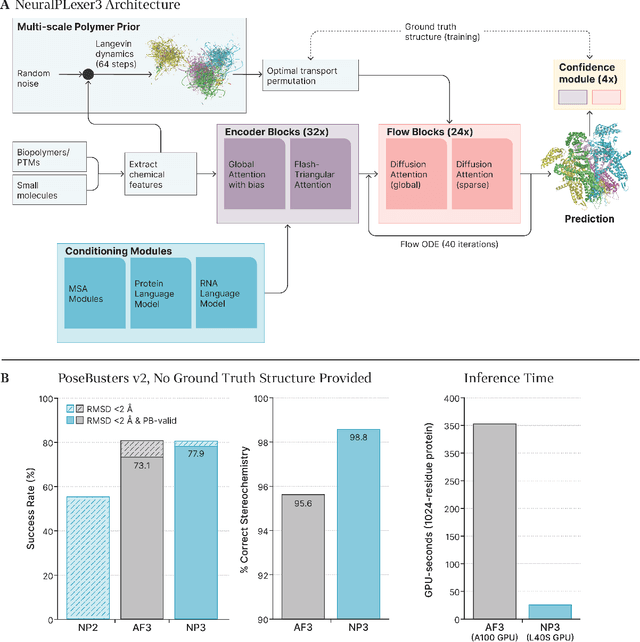
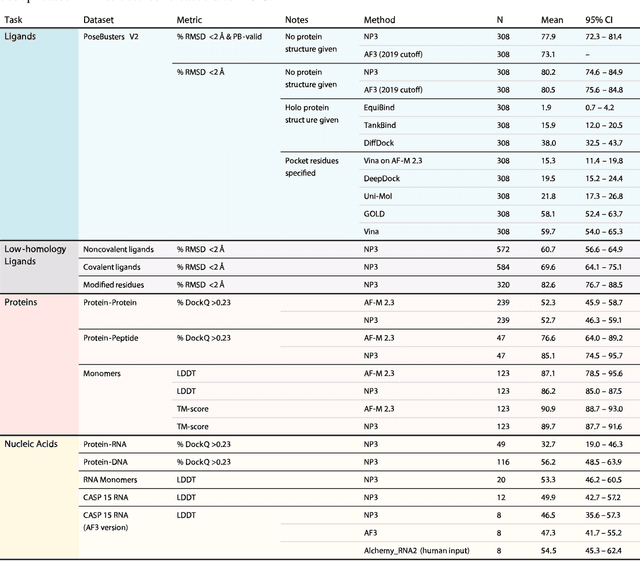
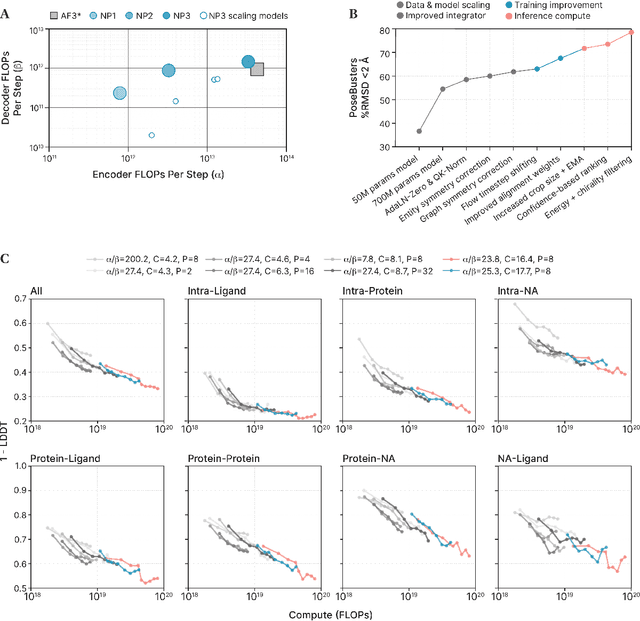
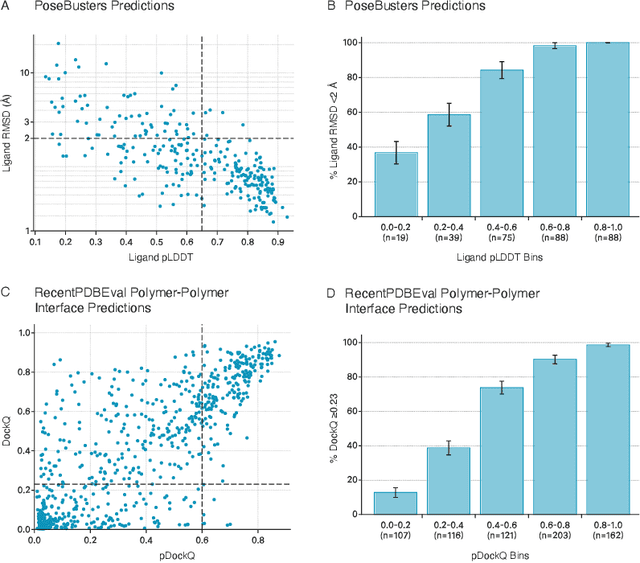
Structure determination is essential to a mechanistic understanding of diseases and the development of novel therapeutics. Machine-learning-based structure prediction methods have made significant advancements by computationally predicting protein and bioassembly structures from sequences and molecular topology alone. Despite substantial progress in the field, challenges remain to deliver structure prediction models to real-world drug discovery. Here, we present NeuralPLexer3 -- a physics-inspired flow-based generative model that achieves state-of-the-art prediction accuracy on key biomolecular interaction types and improves training and sampling efficiency compared to its predecessors and alternative methodologies. Examined through newly developed benchmarking strategies, NeuralPLexer3 excels in vital areas that are crucial to structure-based drug design, such as physical validity and ligand-induced conformational changes.
Multi-modal Molecule Structure-text Model for Text-based Retrieval and Editing
Dec 21, 2022There is increasing adoption of artificial intelligence in drug discovery. However, existing works use machine learning to mainly utilize the chemical structures of molecules yet ignore the vast textual knowledge available in chemistry. Incorporating textual knowledge enables us to realize new drug design objectives, adapt to text-based instructions, and predict complex biological activities. We present a multi-modal molecule structure-text model, MoleculeSTM, by jointly learning molecule's chemical structures and textual descriptions via a contrastive learning strategy. To train MoleculeSTM, we construct the largest multi-modal dataset to date, namely PubChemSTM, with over 280K chemical structure-text pairs. To demonstrate the effectiveness and utility of MoleculeSTM, we design two challenging zero-shot tasks based on text instructions, including structure-text retrieval and molecule editing. MoleculeSTM possesses two main properties: open vocabulary and compositionality via natural language. In experiments, MoleculeSTM obtains the state-of-the-art generalization ability to novel biochemical concepts across various benchmarks.
Dynamic-Backbone Protein-Ligand Structure Prediction with Multiscale Generative Diffusion Models
Sep 30, 2022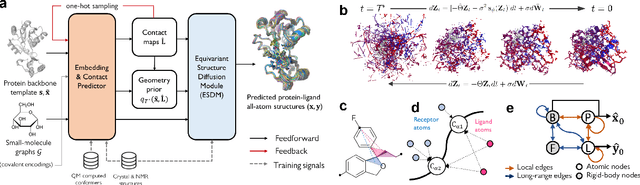
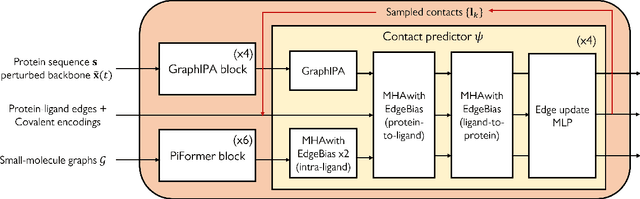
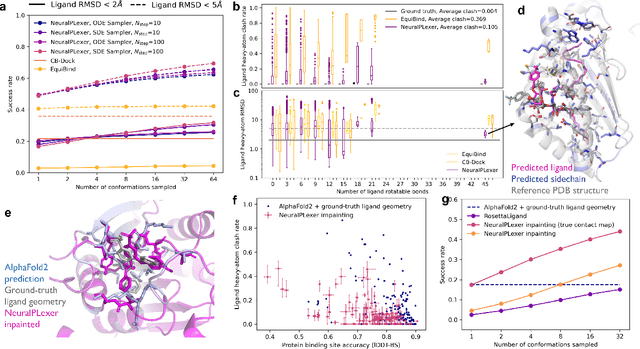
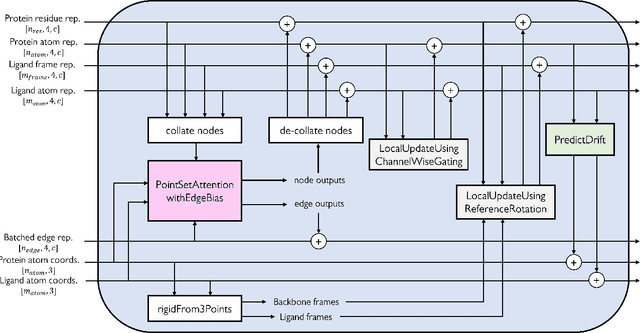
Molecular complexes formed by proteins and small-molecule ligands are ubiquitous, and predicting their 3D structures can facilitate both biological discoveries and the design of novel enzymes or drug molecules. Here we propose NeuralPLexer, a deep generative model framework to rapidly predict protein-ligand complex structures and their fluctuations using protein backbone template and molecular graph inputs. NeuralPLexer jointly samples protein and small-molecule 3D coordinates at an atomistic resolution through a generative model that incorporates biophysical constraints and inferred proximity information into a time-truncated diffusion process. The reverse-time generative diffusion process is learned by a novel stereochemistry-aware equivariant graph transformer that enables efficient, concurrent gradient field prediction for all heavy atoms in the protein-ligand complex. NeuralPLexer outperforms existing physics-based and learning-based methods on benchmarking problems including fixed-backbone blind protein-ligand docking and ligand-coupled binding site repacking. Moreover, we identify preliminary evidence that NeuralPLexer enriches bound-state-like protein structures when applied to systems where protein folding landscapes are significantly altered by the presence of ligands. Our results reveal that a data-driven approach can capture the structural cooperativity among protein and small-molecule entities, showing promise for the computational identification of novel drug targets and the end-to-end differentiable design of functional small-molecules and ligand-binding proteins.
Retrieval-based Controllable Molecule Generation
Aug 23, 2022


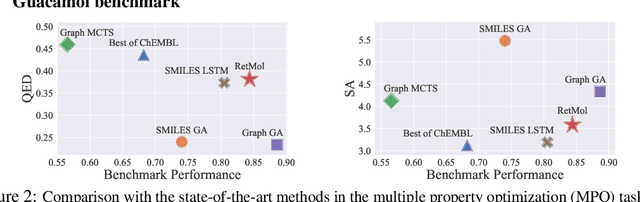
Generating new molecules with specified chemical and biological properties via generative models has emerged as a promising direction for drug discovery. However, existing methods require extensive training/fine-tuning with a large dataset, often unavailable in real-world generation tasks. In this work, we propose a new retrieval-based framework for controllable molecule generation. We use a small set of exemplar molecules, i.e., those that (partially) satisfy the design criteria, to steer the pre-trained generative model towards synthesizing molecules that satisfy the given design criteria. We design a retrieval mechanism that retrieves and fuses the exemplar molecules with the input molecule, which is trained by a new self-supervised objective that predicts the nearest neighbor of the input molecule. We also propose an iterative refinement process to dynamically update the generated molecules and retrieval database for better generalization. Our approach is agnostic to the choice of generative models and requires no task-specific fine-tuning. On various tasks ranging from simple design criteria to a challenging real-world scenario for designing lead compounds that bind to the SARS-CoV-2 main protease, we demonstrate our approach extrapolates well beyond the retrieval database, and achieves better performance and wider applicability than previous methods.
UNiTE: Unitary N-body Tensor Equivariant Network with Applications to Quantum Chemistry
Jun 06, 2021
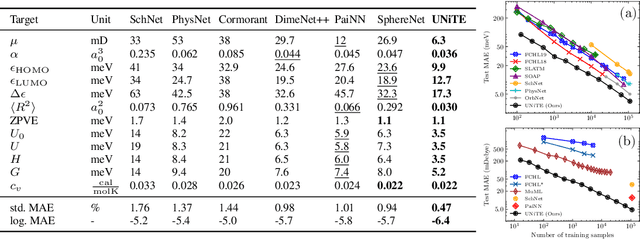
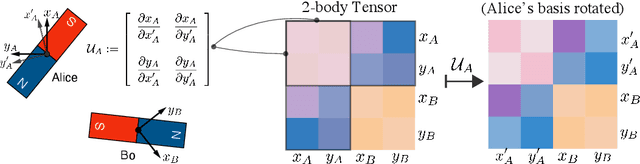
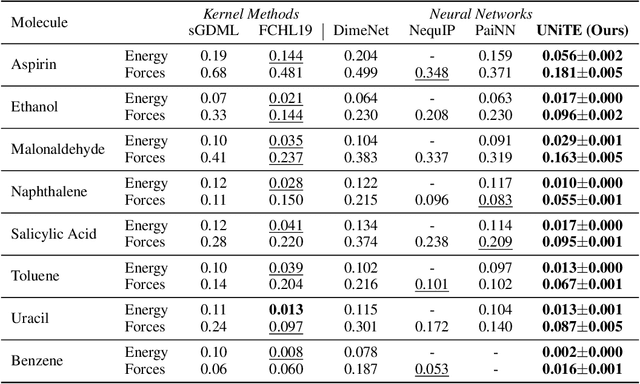
Equivariant neural networks have been successful in incorporating various types of symmetries, but are mostly limited to vector representations of geometric objects. Despite the prevalence of higher-order tensors in various application domains, e.g. in quantum chemistry, equivariant neural networks for general tensors remain unexplored. Previous strategies for learning equivariant functions on tensors mostly rely on expensive tensor factorization which is not scalable when the dimensionality of the problem becomes large. In this work, we propose unitary $N$-body tensor equivariant neural network (UNiTE), an architecture for a general class of symmetric tensors called $N$-body tensors. The proposed neural network is equivariant with respect to the actions of a unitary group, such as the group of 3D rotations. Furthermore, it has a linear time complexity with respect to the number of non-zero elements in the tensor. We also introduce a normalization method, viz., Equivariant Normalization, to improve generalization of the neural network while preserving symmetry. When applied to quantum chemistry, UNiTE outperforms all state-of-the-art machine learning methods of that domain with over 110% average improvements on multiple benchmarks. Finally, we show that UNiTE achieves a robust zero-shot generalization performance on diverse down stream chemistry tasks, while being three orders of magnitude faster than conventional numerical methods with competitive accuracy.
Multi-task learning for electronic structure to predict and explore molecular potential energy surfaces
Nov 11, 2020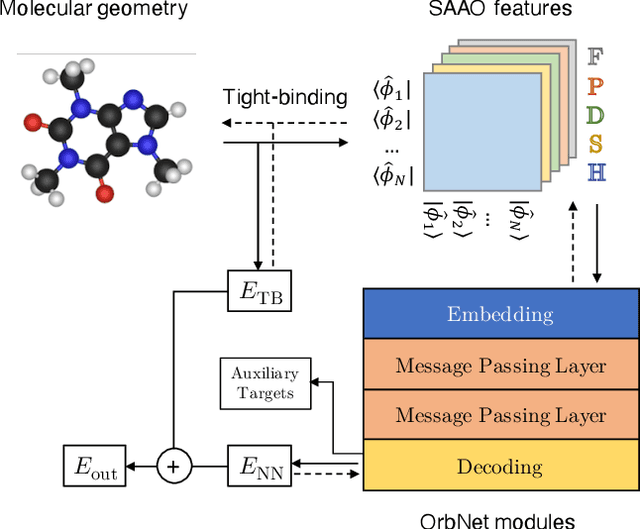

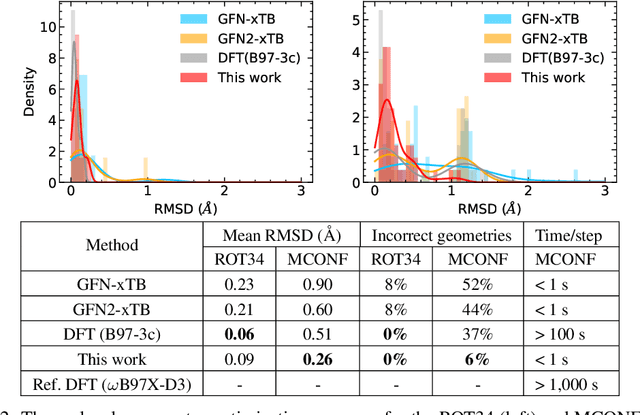
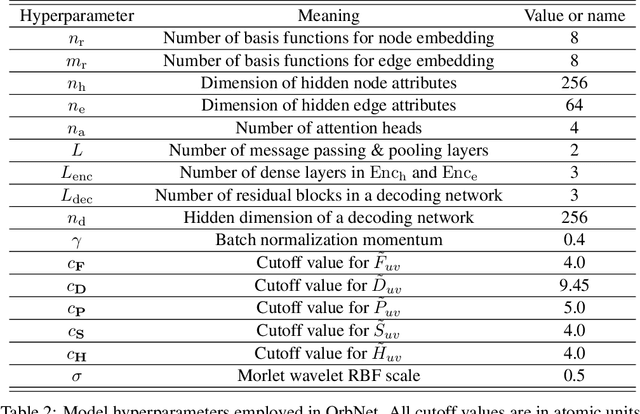
We refine the OrbNet model to accurately predict energy, forces, and other response properties for molecules using a graph neural-network architecture based on features from low-cost approximated quantum operators in the symmetry-adapted atomic orbital basis. The model is end-to-end differentiable due to the derivation of analytic gradients for all electronic structure terms, and is shown to be transferable across chemical space due to the use of domain-specific features. The learning efficiency is improved by incorporating physically motivated constraints on the electronic structure through multi-task learning. The model outperforms existing methods on energy prediction tasks for the QM9 dataset and for molecular geometry optimizations on conformer datasets, at a computational cost that is thousand-fold or more reduced compared to conventional quantum-chemistry calculations (such as density functional theory) that offer similar accuracy.
OrbNet: Deep Learning for Quantum Chemistry Using Symmetry-Adapted Atomic-Orbital Features
Jul 15, 2020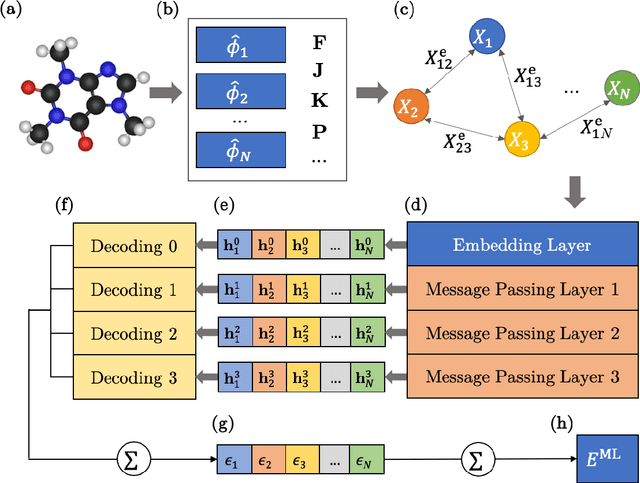
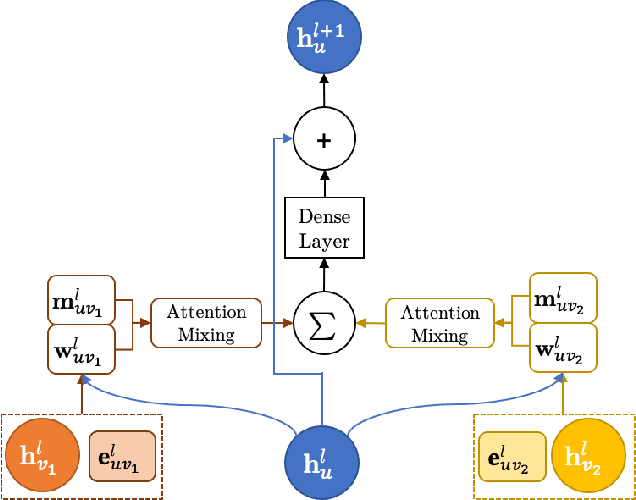
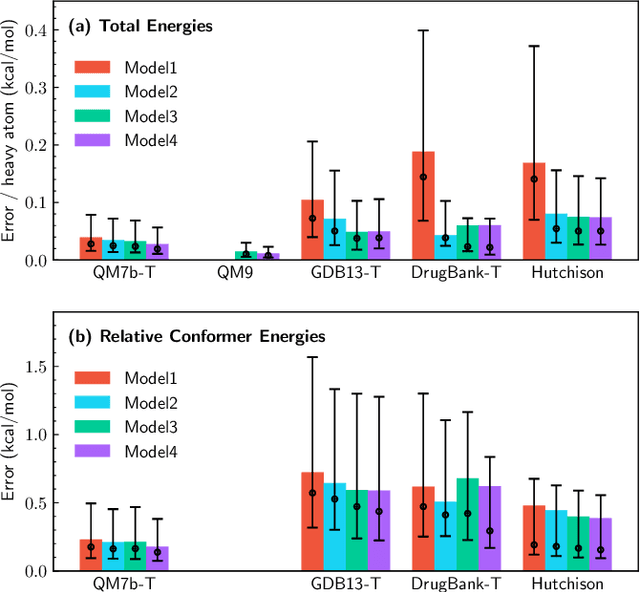
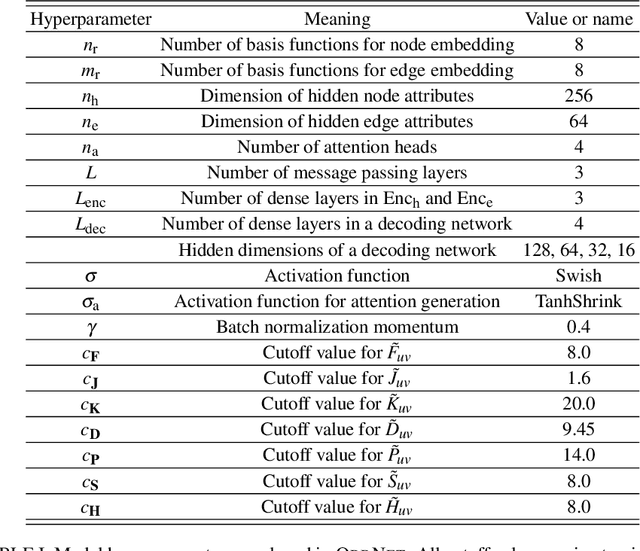
We introduce a machine learning method in which energy solutions from the Schrodinger equation are predicted using symmetry adapted atomic orbitals features and a graph neural-network architecture. \textsc{OrbNet} is shown to outperform existing methods in terms of learning efficiency and transferability for the prediction of density functional theory results while employing low-cost features that are obtained from semi-empirical electronic structure calculations. For applications to datasets of drug-like molecules, including QM7b-T, QM9, GDB-13-T, DrugBank, and the conformer benchmark dataset of Folmsbee and Hutchison, \textsc{OrbNet} predicts energies within chemical accuracy of DFT at a computational cost that is thousand-fold or more reduced.