 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-task learning for electronic structure to predict and explore molecular potential energy surfaces
Nov 11, 2020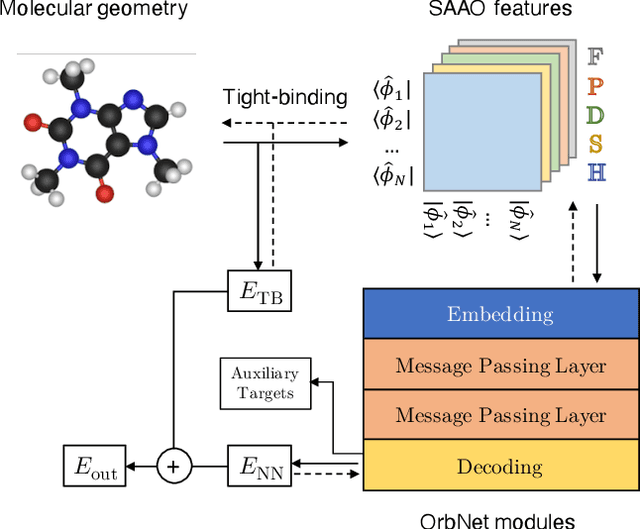

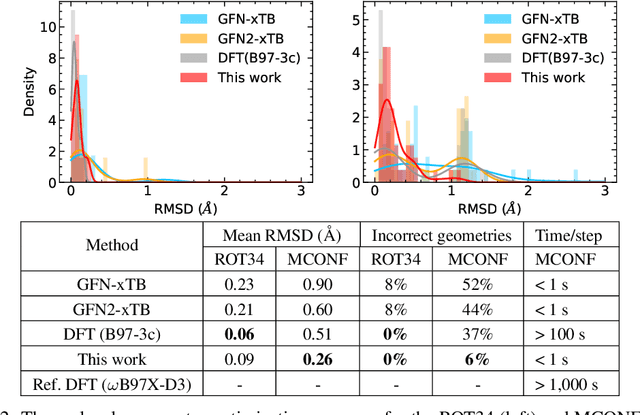
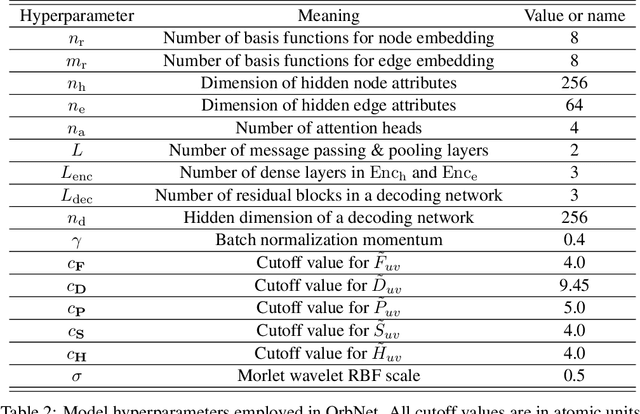
We refine the OrbNet model to accurately predict energy, forces, and other response properties for molecules using a graph neural-network architecture based on features from low-cost approximated quantum operators in the symmetry-adapted atomic orbital basis. The model is end-to-end differentiable due to the derivation of analytic gradients for all electronic structure terms, and is shown to be transferable across chemical space due to the use of domain-specific features. The learning efficiency is improved by incorporating physically motivated constraints on the electronic structure through multi-task learning. The model outperforms existing methods on energy prediction tasks for the QM9 dataset and for molecular geometry optimizations on conformer datasets, at a computational cost that is thousand-fold or more reduced compared to conventional quantum-chemistry calculations (such as density functional theory) that offer similar accuracy.