 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOrbNet: Deep Learning for Quantum Chemistry Using Symmetry-Adapted Atomic-Orbital Features
Paper and Code
Jul 15, 2020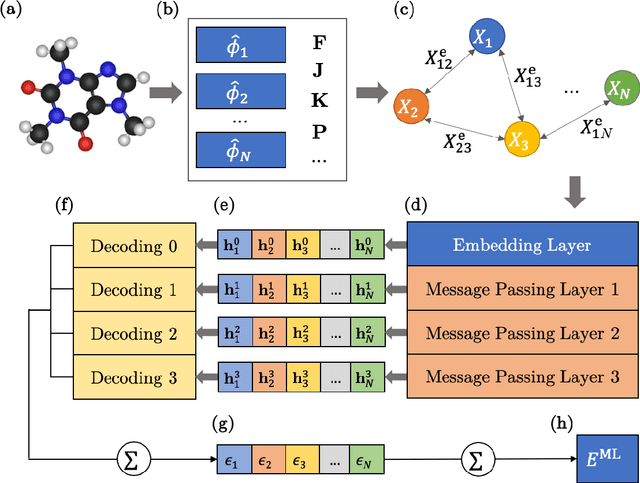
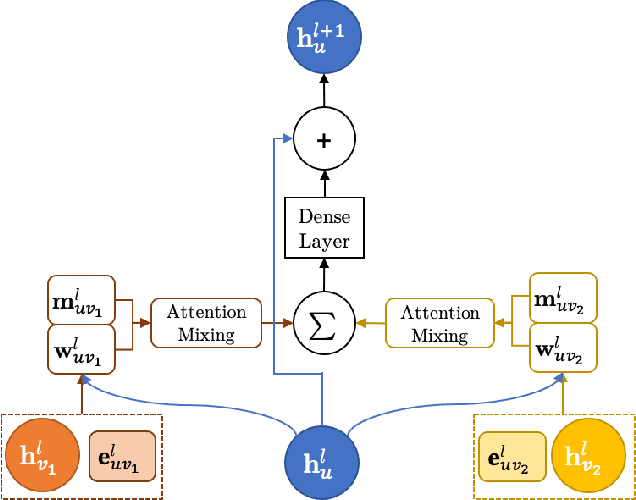
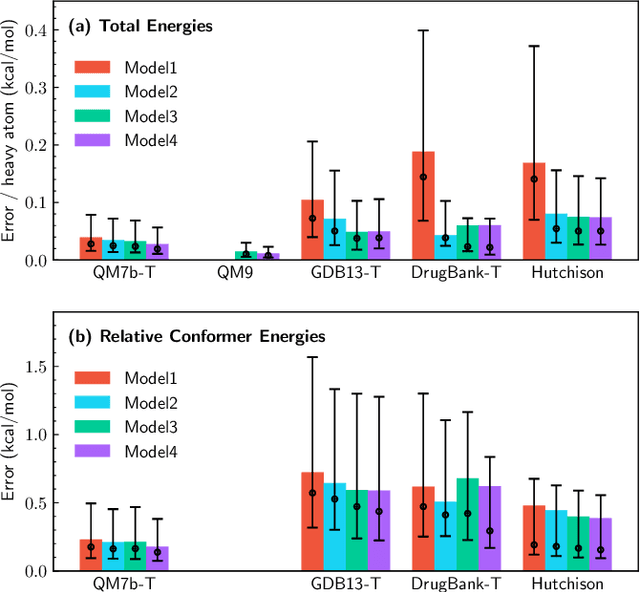
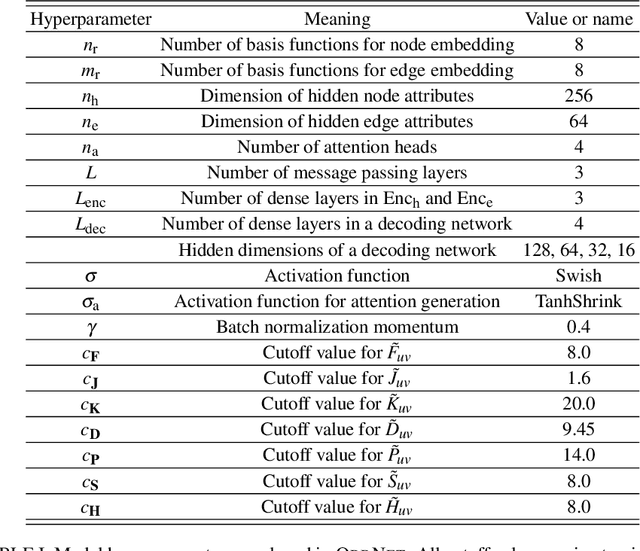
We introduce a machine learning method in which energy solutions from the Schrodinger equation are predicted using symmetry adapted atomic orbitals features and a graph neural-network architecture. \textsc{OrbNet} is shown to outperform existing methods in terms of learning efficiency and transferability for the prediction of density functional theory results while employing low-cost features that are obtained from semi-empirical electronic structure calculations. For applications to datasets of drug-like molecules, including QM7b-T, QM9, GDB-13-T, DrugBank, and the conformer benchmark dataset of Folmsbee and Hutchison, \textsc{OrbNet} predicts energies within chemical accuracy of DFT at a computational cost that is thousand-fold or more reduced.