 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDynamic-Backbone Protein-Ligand Structure Prediction with Multiscale Generative Diffusion Models
Paper and Code
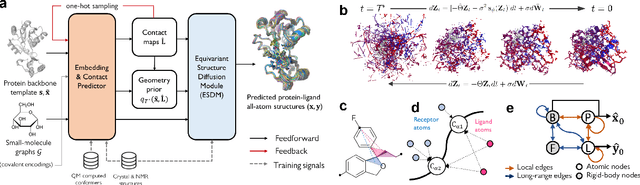
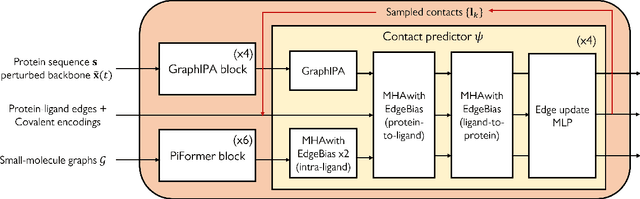
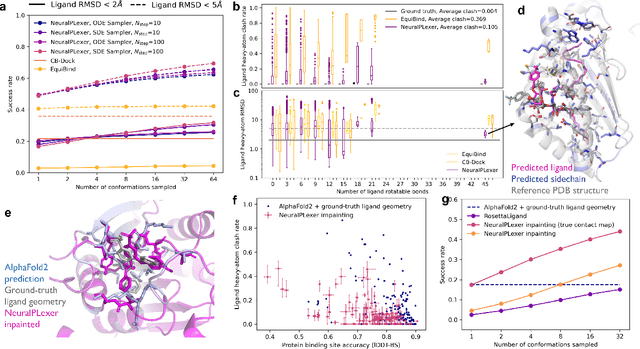
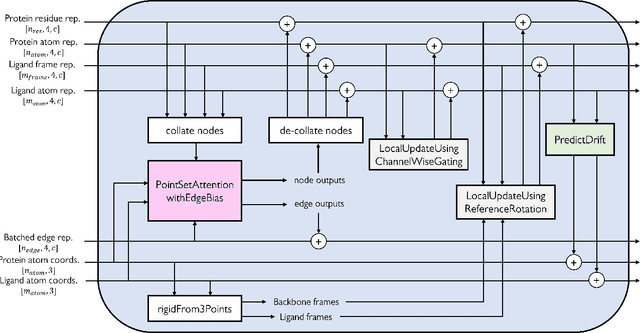
Molecular complexes formed by proteins and small-molecule ligands are ubiquitous, and predicting their 3D structures can facilitate both biological discoveries and the design of novel enzymes or drug molecules. Here we propose NeuralPLexer, a deep generative model framework to rapidly predict protein-ligand complex structures and their fluctuations using protein backbone template and molecular graph inputs. NeuralPLexer jointly samples protein and small-molecule 3D coordinates at an atomistic resolution through a generative model that incorporates biophysical constraints and inferred proximity information into a time-truncated diffusion process. The reverse-time generative diffusion process is learned by a novel stereochemistry-aware equivariant graph transformer that enables efficient, concurrent gradient field prediction for all heavy atoms in the protein-ligand complex. NeuralPLexer outperforms existing physics-based and learning-based methods on benchmarking problems including fixed-backbone blind protein-ligand docking and ligand-coupled binding site repacking. Moreover, we identify preliminary evidence that NeuralPLexer enriches bound-state-like protein structures when applied to systems where protein folding landscapes are significantly altered by the presence of ligands. Our results reveal that a data-driven approach can capture the structural cooperativity among protein and small-molecule entities, showing promise for the computational identification of novel drug targets and the end-to-end differentiable design of functional small-molecules and ligand-binding proteins.