 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGlobal Context Enhanced Graph Neural Networks for Session-based Recommendation
Jun 09, 2021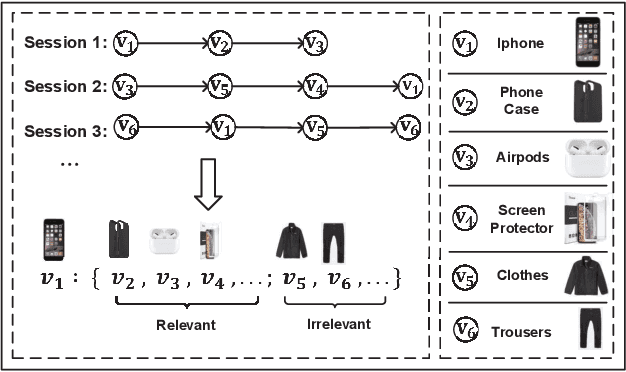
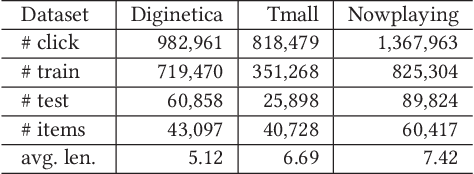
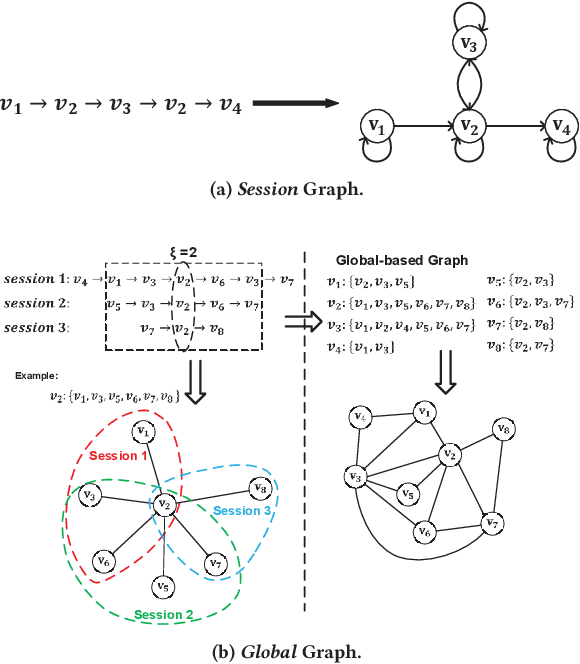
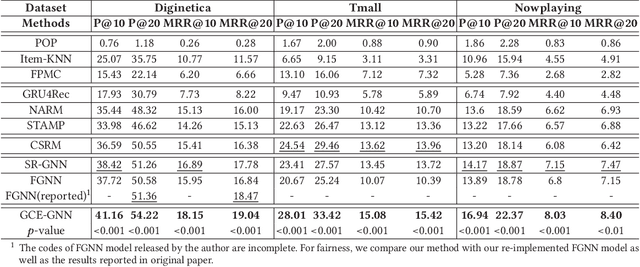
Session-based recommendation (SBR) is a challenging task, which aims at recommending items based on anonymous behavior sequences. Almost all the existing solutions for SBR model user preference only based on the current session without exploiting the other sessions, which may contain both relevant and irrelevant item-transitions to the current session. This paper proposes a novel approach, called Global Context Enhanced Graph Neural Networks (GCE-GNN) to exploit item transitions over all sessions in a more subtle manner for better inferring the user preference of the current session. Specifically, GCE-GNN learns two levels of item embeddings from session graph and global graph, respectively: (i) Session graph, which is to learn the session-level item embedding by modeling pairwise item-transitions within the current session; and (ii) Global graph, which is to learn the global-level item embedding by modeling pairwise item-transitions over all sessions. In GCE-GNN, we propose a novel global-level item representation learning layer, which employs a session-aware attention mechanism to recursively incorporate the neighbors' embeddings of each node on the global graph. We also design a session-level item representation learning layer, which employs a GNN on the session graph to learn session-level item embeddings within the current session. Moreover, GCE-GNN aggregates the learnt item representations in the two levels with a soft attention mechanism. Experiments on three benchmark datasets demonstrate that GCE-GNN outperforms the state-of-the-art methods consistently.
* arXiv admin note: substantial text overlap with arXiv:2011.10173
Recent Advances in Network-based Methods for Disease Gene Prediction
Jul 19, 2020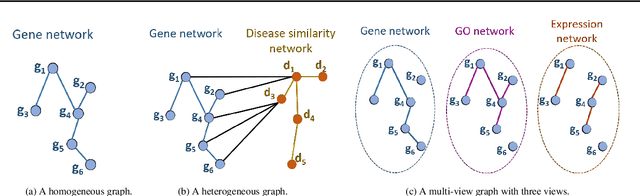
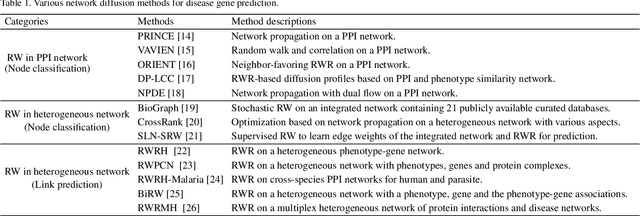
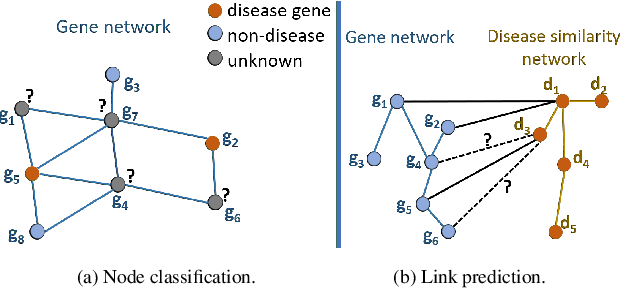

Disease-gene association through Genome-wide association study (GWAS) is an arduous task for researchers. Investigating single nucleotide polymorphisms (SNPs) that correlate with specific diseases needs statistical analysis of associations, considering the huge number of possible disease mutations. The most important drawback of GWAS analysis in addition to its high cost is the large number of false-positives. Thus, researchers search for more evidence to cross-check their results through different sources. To provide the researchers with alternative low-cost disease-gene association evidence, computational approaches come into play. Since molecular networks are able to capture complex interplay among molecules in diseases, they become one of the most extensively used data for disease-gene association prediction. In this survey, we aim to provide a comprehensive and an up-to-date review of network-based methods for disease gene prediction. We also conduct an empirical analysis on 14 state-of-the-art methods. To summarize, we first elucidate the task definition for disease gene prediction. Secondly, we categorize existing network-based efforts into network diffusion methods, traditional machine learning methods with handcrafted graph features and graph representation learning methods. Thirdly, an empirical analysis is conducted to evaluate the performance of the selected methods across seven diseases. We also provide distinguishing findings about the discussed methods based on our empirical analysis. Finally, we highlight potential research directions for future studies on disease gene prediction.
SL$^2$MF: Predicting Synthetic Lethality in Human Cancers via Logistic Matrix Factorization
Oct 20, 2018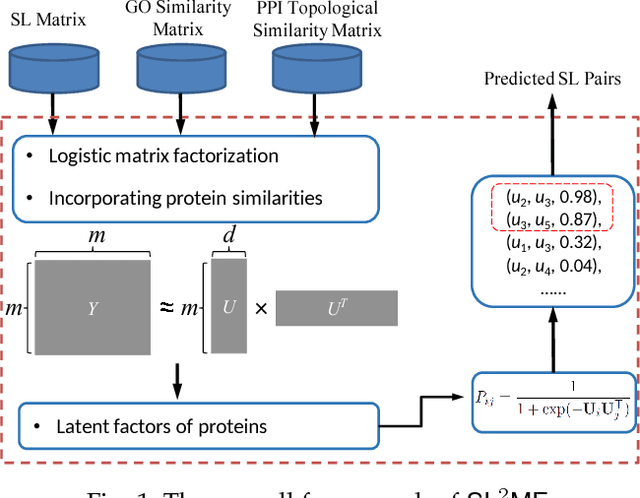
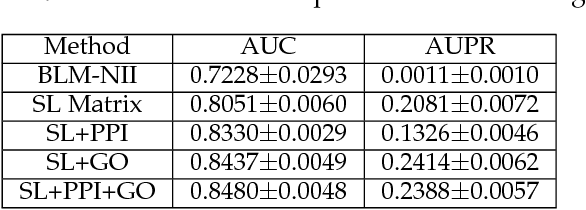
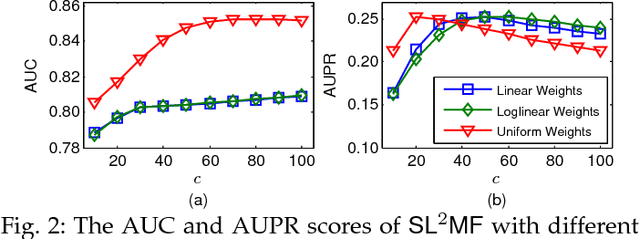

Synthetic lethality (SL) is a promising concept for novel discovery of anti-cancer drug targets. However, wet-lab experiments for detecting SLs are faced with various challenges, such as high cost, low consistency across platforms or cell lines. Therefore, computational prediction methods are needed to address these issues. This paper proposes a novel SL prediction method, named SL2MF, which employs logistic matrix factorization to learn latent representations of genes from the observed SL data. The probability that two genes are likely to form SL is modeled by the linear combination of gene latent vectors. As known SL pairs are more trustworthy than unknown pairs, we design importance weighting schemes to assign higher importance weights for known SL pairs and lower importance weights for unknown pairs in SL2MF. Moreover, we also incorporate biological knowledge about genes from protein-protein interaction (PPI) data and Gene Ontology (GO). In particular, we calculate the similarity between genes based on their GO annotations and topological properties in the PPI network. Extensive experiments on the SL interaction data from SynLethDB database have been conducted to demonstrate the effectiveness of SL2MF.
Hyperbolic Recommender Systems
Sep 05, 2018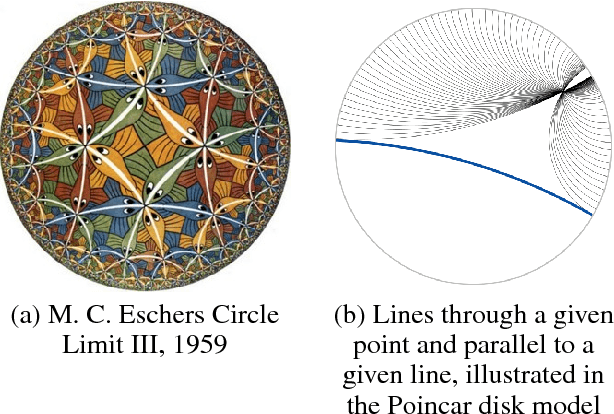
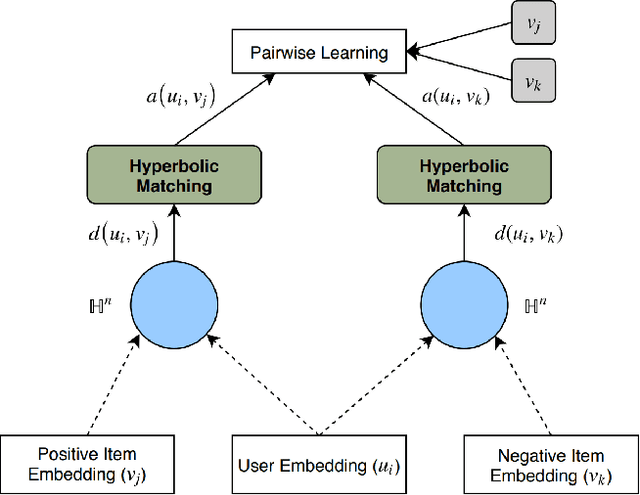
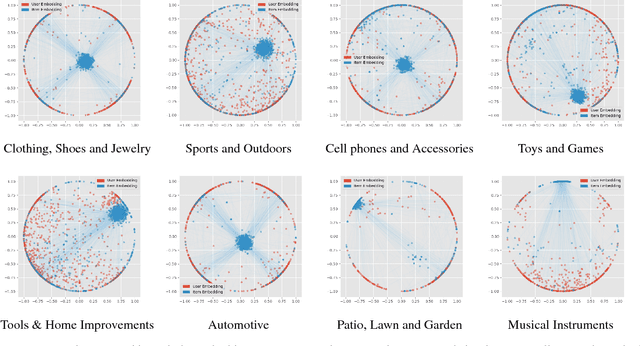
Many well-established recommender systems are based on representation learning in Euclidean space. In these models, matching functions such as the Euclidean distance or inner product are typically used for computing similarity scores between user and item embeddings. This paper investigates the notion of learning user and item representations in Hyperbolic space. In this paper, we argue that Hyperbolic space is more suitable for learning user-item embeddings in the recommendation domain. Unlike Euclidean spaces, Hyperbolic spaces are intrinsically equipped to handle hierarchical structure, encouraged by its property of exponentially increasing distances away from origin. We propose HyperBPR (Hyperbolic Bayesian Personalized Ranking), a conceptually simple but highly effective model for the task at hand. Our proposed HyperBPR not only outperforms their Euclidean counterparts, but also achieves state-of-the-art performance on multiple benchmark datasets, demonstrating the effectiveness of personalized recommendation in Hyperbolic space.
Attention-based Group Recommendation
Jul 10, 2018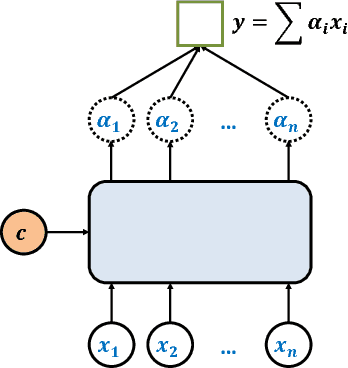
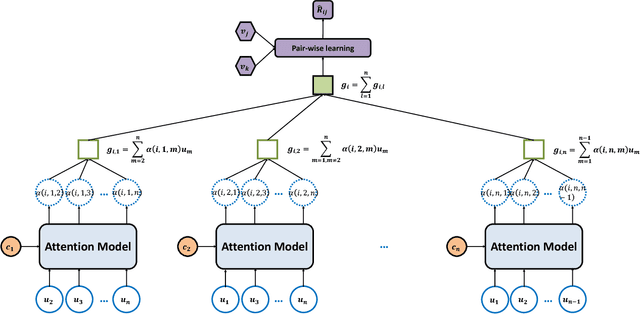
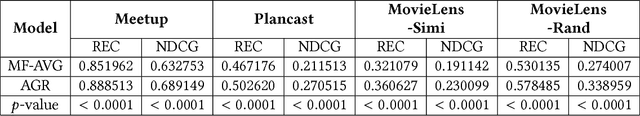
Group recommendation aims to recommend items for a group of users, e.g., recommending a restaurant for a group of colleagues. The group recommendation problem is challenging, in that a good model should understand the group decision making process appropriately: users are likely to follow decisions of only a few users, who are group's leaders or experts. To address this challenge, we propose using an attention mechanism to capture the impact of each user in a group. Specifically, our model learns the influence weight of each user in a group and recommends items to the group based on its members' weighted preferences. Moreover, our model can dynamically adjust the weight of each user across the groups; thus, the model provides a new and flexible method to model the complicated group decision making process, which differentiates us from other existing solutions. Through extensive experiments, it has demonstrated that our model significantly outperforms baseline methods for the group recommendation problem.
Adversarial adaptive 1-D convolutional neural networks for bearing fault diagnosis under varying working condition
May 09, 2018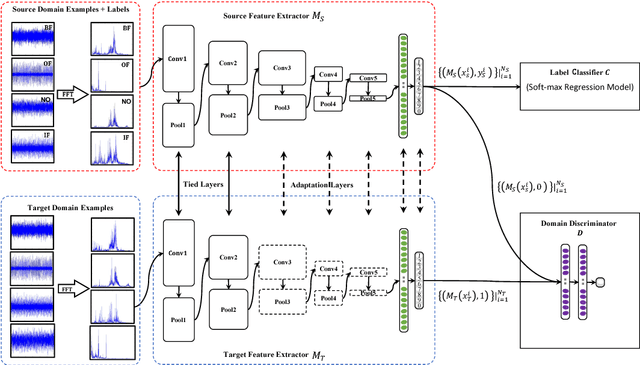
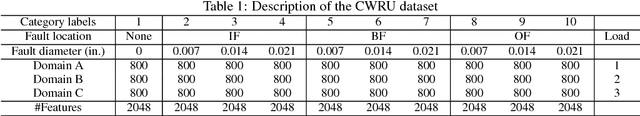
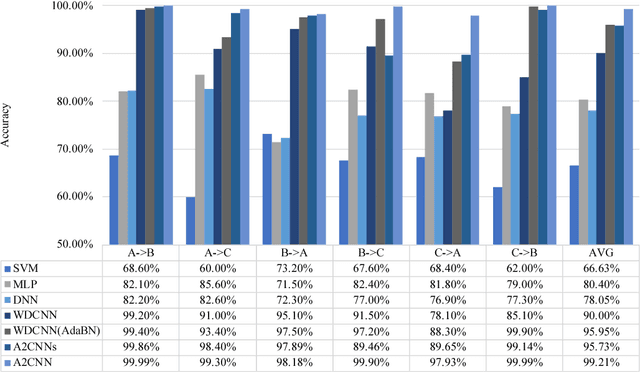
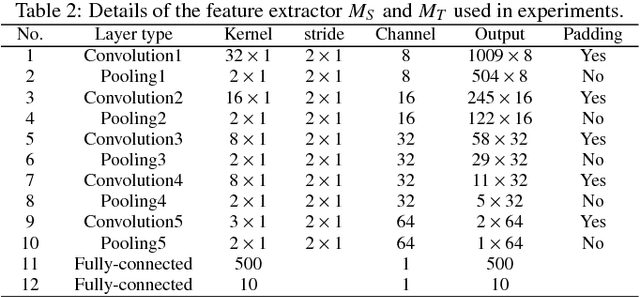
Traditional intelligent fault diagnosis of rolling bearings work well only under a common assumption that the labeled training data (source domain) and unlabeled testing data (target domain) are drawn from the same distribution. However, in many real-world applications, this assumption does not hold, especially when the working condition varies. In this paper, a new adversarial adaptive 1-D CNN called A2CNN is proposed to address this problem. A2CNN consists of four parts, namely, a source feature extractor, a target feature extractor, a label classifier and a domain discriminator. The layers between the source and target feature extractor are partially untied during the training stage to take both training efficiency and domain adaptation into consideration. Experiments show that A2CNN has strong fault-discriminative and domain-invariant capacity, and therefore can achieve high accuracy under different working conditions. We also visualize the learned features and the networks to explore the reasons behind the high performance of our proposed model.
Classification and its applications for drug-target interaction identification
Mar 12, 2015
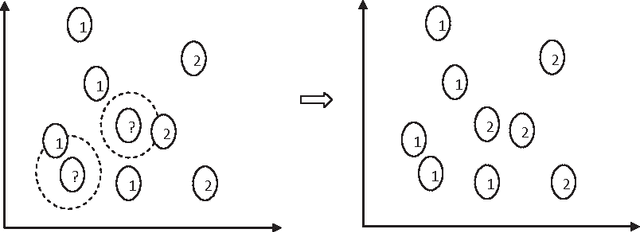

Classification is one of the most popular and widely used supervised learning tasks, which categorizes objects into predefined classes based on known knowledge. Classification has been an important research topic in machine learning and data mining. Different classification methods have been proposed and applied to deal with various real-world problems. Unlike unsupervised learning such as clustering, a classifier is typically trained with labeled data before being used to make prediction, and usually achieves higher accuracy than unsupervised one. In this paper, we first define classification and then review several representative methods. After that, we study in details the application of classification to a critical problem in drug discovery, i.e., drug-target prediction, due to the challenges in predicting possible interactions between drugs and targets.
Microbial community pattern detection in human body habitats via ensemble clustering framework
Jan 04, 2015
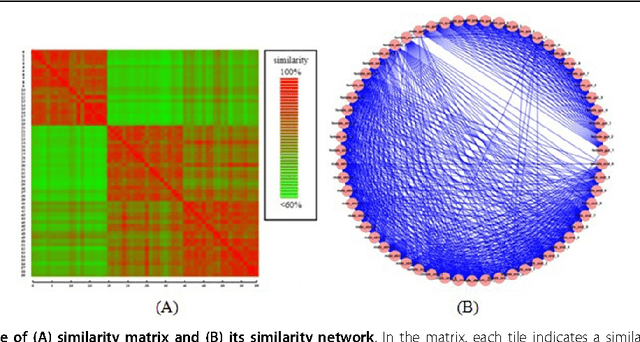
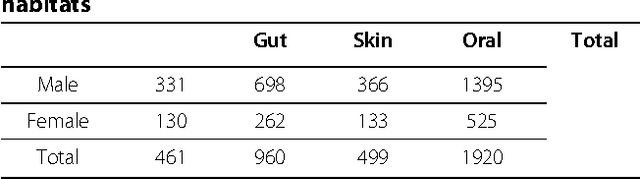
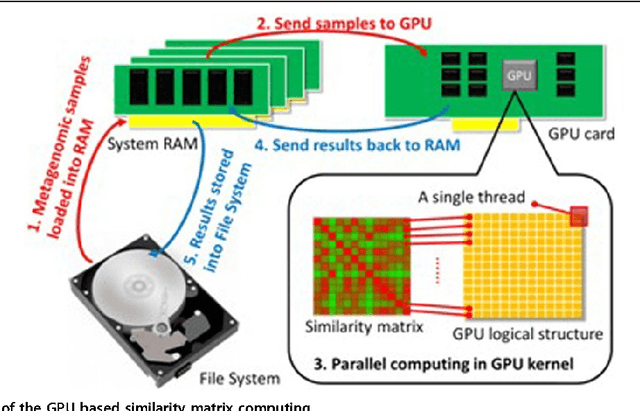
The human habitat is a host where microbial species evolve, function, and continue to evolve. Elucidating how microbial communities respond to human habitats is a fundamental and critical task, as establishing baselines of human microbiome is essential in understanding its role in human disease and health. However, current studies usually overlook a complex and interconnected landscape of human microbiome and limit the ability in particular body habitats with learning models of specific criterion. Therefore, these methods could not capture the real-world underlying microbial patterns effectively. To obtain a comprehensive view, we propose a novel ensemble clustering framework to mine the structure of microbial community pattern on large-scale metagenomic data. Particularly, we first build a microbial similarity network via integrating 1920 metagenomic samples from three body habitats of healthy adults. Then a novel symmetric Nonnegative Matrix Factorization (NMF) based ensemble model is proposed and applied onto the network to detect clustering pattern. Extensive experiments are conducted to evaluate the effectiveness of our model on deriving microbial community with respect to body habitat and host gender. From clustering results, we observed that body habitat exhibits a strong bound but non-unique microbial structural patterns. Meanwhile, human microbiome reveals different degree of structural variations over body habitat and host gender. In summary, our ensemble clustering framework could efficiently explore integrated clustering results to accurately identify microbial communities, and provide a comprehensive view for a set of microbial communities. Such trends depict an integrated biography of microbial communities, which offer a new insight towards uncovering pathogenic model of human microbiome.
* BMC Systems Biology 2014