 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeETDock: A Novel Equivariant Transformer for Protein-Ligand Docking
Oct 12, 2023



Predicting the docking between proteins and ligands is a crucial and challenging task for drug discovery. However, traditional docking methods mainly rely on scoring functions, and deep learning-based docking approaches usually neglect the 3D spatial information of proteins and ligands, as well as the graph-level features of ligands, which limits their performance. To address these limitations, we propose an equivariant transformer neural network for protein-ligand docking pose prediction. Our approach involves the fusion of ligand graph-level features by feature processing, followed by the learning of ligand and protein representations using our proposed TAMformer module. Additionally, we employ an iterative optimization approach based on the predicted distance matrix to generate refined ligand poses. The experimental results on real datasets show that our model can achieve state-of-the-art performance.
Predicting Protein-Ligand Binding Affinity with Equivariant Line Graph Network
Oct 27, 2022



Binding affinity prediction of three-dimensional (3D) protein ligand complexes is critical for drug repositioning and virtual drug screening. Existing approaches transform a 3D protein-ligand complex to a two-dimensional (2D) graph, and then use graph neural networks (GNNs) to predict its binding affinity. However, the node and edge features of the 2D graph are extracted based on invariant local coordinate systems of the 3D complex. As a result, the method can not fully learn the global information of the complex, such as, the physical symmetry and the topological information of bonds. To address these issues, we propose a novel Equivariant Line Graph Network (ELGN) for affinity prediction of 3D protein ligand complexes. The proposed ELGN firstly adds a super node to the 3D complex, and then builds a line graph based on the 3D complex. After that, ELGN uses a new E(3)-equivariant network layer to pass the messages between nodes and edges based on the global coordinate system of the 3D complex. Experimental results on two real datasets demonstrate the effectiveness of ELGN over several state-of-the-art baselines.
MARS: A Motif-based Autoregressive Model for Retrosynthesis Prediction
Sep 27, 2022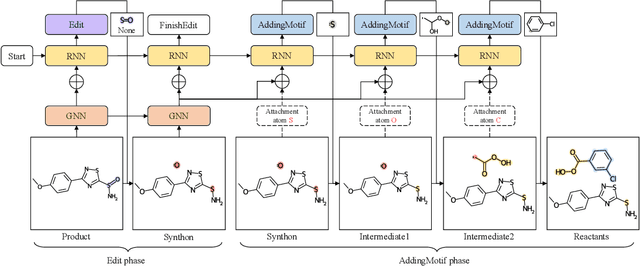
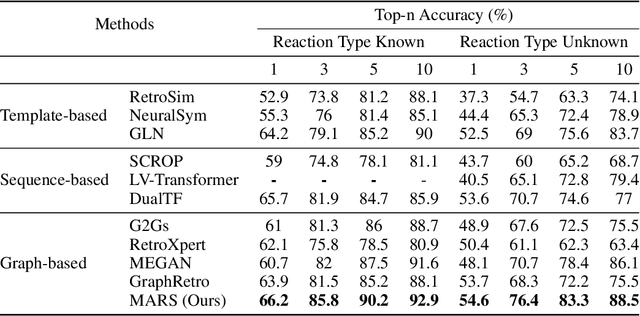
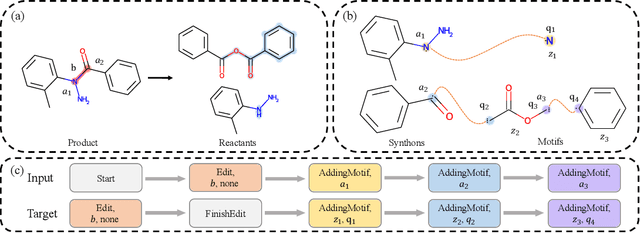
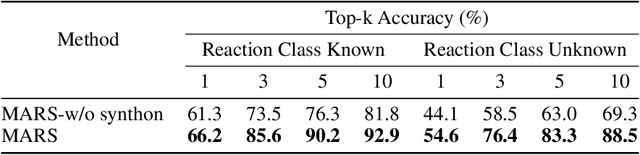
Retrosynthesis is a major task for drug discovery. It is formulated as a graph-generating problem by many existing approaches. Specifically, these methods firstly identify the reaction center, and break target molecule accordingly to generate synthons. Reactants are generated by either adding atoms sequentially to synthon graphs or directly adding proper leaving groups. However, both two strategies suffer since adding atoms results in a long prediction sequence which increases generation difficulty, while adding leaving groups can only consider the ones in the training set which results in poor generalization. In this paper, we propose a novel end-to-end graph generation model for retrosynthesis prediction, which sequentially identifies the reaction center, generates the synthons, and adds motifs to the synthons to generate reactants. Since chemically meaningful motifs are bigger than atoms and smaller than leaving groups, our method enjoys lower prediction complexity than adding atoms and better generalization than adding leaving groups. Experiments on a benchmark dataset show that the proposed model significantly outperforms previous state-of-the-art algorithms.
Recent Advances in Network-based Methods for Disease Gene Prediction
Jul 19, 2020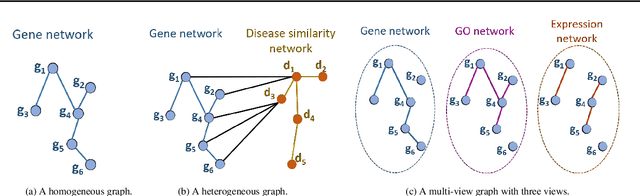
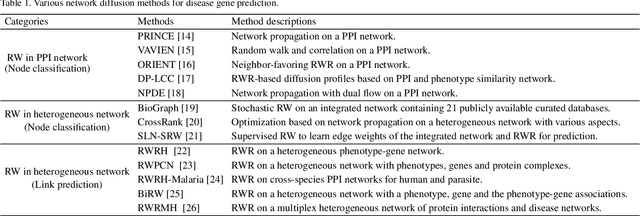
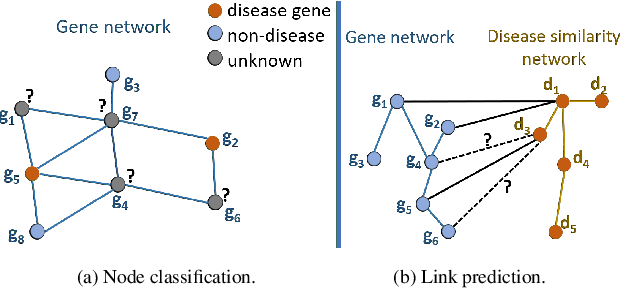

Disease-gene association through Genome-wide association study (GWAS) is an arduous task for researchers. Investigating single nucleotide polymorphisms (SNPs) that correlate with specific diseases needs statistical analysis of associations, considering the huge number of possible disease mutations. The most important drawback of GWAS analysis in addition to its high cost is the large number of false-positives. Thus, researchers search for more evidence to cross-check their results through different sources. To provide the researchers with alternative low-cost disease-gene association evidence, computational approaches come into play. Since molecular networks are able to capture complex interplay among molecules in diseases, they become one of the most extensively used data for disease-gene association prediction. In this survey, we aim to provide a comprehensive and an up-to-date review of network-based methods for disease gene prediction. We also conduct an empirical analysis on 14 state-of-the-art methods. To summarize, we first elucidate the task definition for disease gene prediction. Secondly, we categorize existing network-based efforts into network diffusion methods, traditional machine learning methods with handcrafted graph features and graph representation learning methods. Thirdly, an empirical analysis is conducted to evaluate the performance of the selected methods across seven diseases. We also provide distinguishing findings about the discussed methods based on our empirical analysis. Finally, we highlight potential research directions for future studies on disease gene prediction.
Semantic Hierarchy Preserving Deep Hashing for Large-scale Image Retrieval
Apr 12, 2019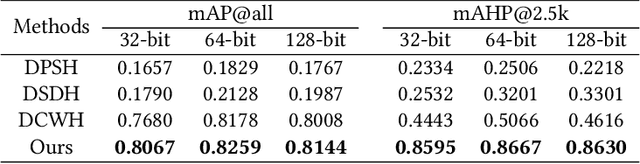

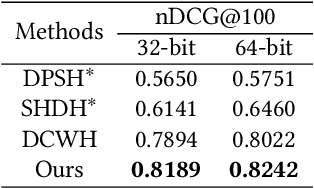
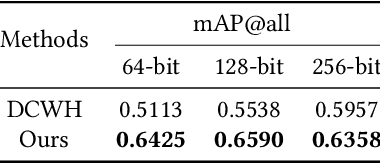
Convolutional neural networks have been widely used in content-based image retrieval. To better deal with large-scale data, the deep hashing model is proposed as an effective method, which maps an image to a binary code that can be used for hashing search. However, most existing deep hashing models only utilize fine-level semantic labels or convert them to similar/dissimilar labels for training. The natural semantic hierarchy structures are ignored in the training stage of the deep hashing model. In this paper, we present an effective algorithm to train a deep hashing model that can preserve a semantic hierarchy structure for large-scale image retrieval. Experiments on two datasets show that our method improves the fine-level retrieval performance. Meanwhile, our model achieves state-of-the-art results in terms of hierarchical retrieval.
Microbial community pattern detection in human body habitats via ensemble clustering framework
Jan 04, 2015
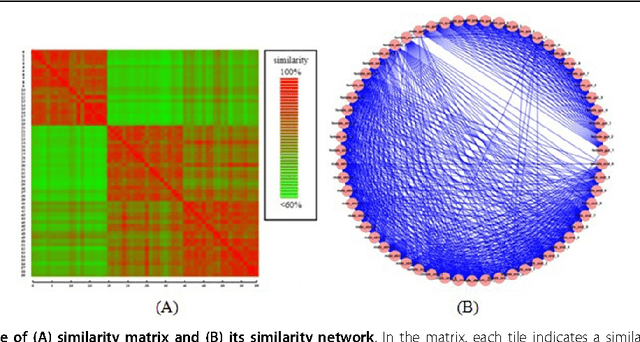
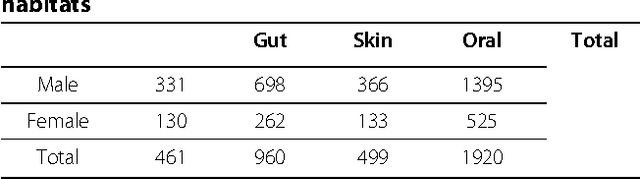
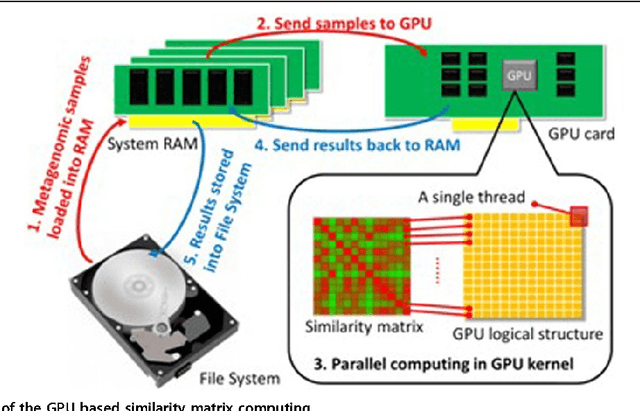
The human habitat is a host where microbial species evolve, function, and continue to evolve. Elucidating how microbial communities respond to human habitats is a fundamental and critical task, as establishing baselines of human microbiome is essential in understanding its role in human disease and health. However, current studies usually overlook a complex and interconnected landscape of human microbiome and limit the ability in particular body habitats with learning models of specific criterion. Therefore, these methods could not capture the real-world underlying microbial patterns effectively. To obtain a comprehensive view, we propose a novel ensemble clustering framework to mine the structure of microbial community pattern on large-scale metagenomic data. Particularly, we first build a microbial similarity network via integrating 1920 metagenomic samples from three body habitats of healthy adults. Then a novel symmetric Nonnegative Matrix Factorization (NMF) based ensemble model is proposed and applied onto the network to detect clustering pattern. Extensive experiments are conducted to evaluate the effectiveness of our model on deriving microbial community with respect to body habitat and host gender. From clustering results, we observed that body habitat exhibits a strong bound but non-unique microbial structural patterns. Meanwhile, human microbiome reveals different degree of structural variations over body habitat and host gender. In summary, our ensemble clustering framework could efficiently explore integrated clustering results to accurately identify microbial communities, and provide a comprehensive view for a set of microbial communities. Such trends depict an integrated biography of microbial communities, which offer a new insight towards uncovering pathogenic model of human microbiome.
* BMC Systems Biology 2014