 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMARS: A Motif-based Autoregressive Model for Retrosynthesis Prediction
Sep 27, 2022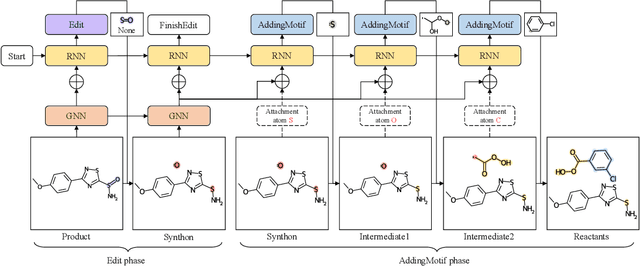
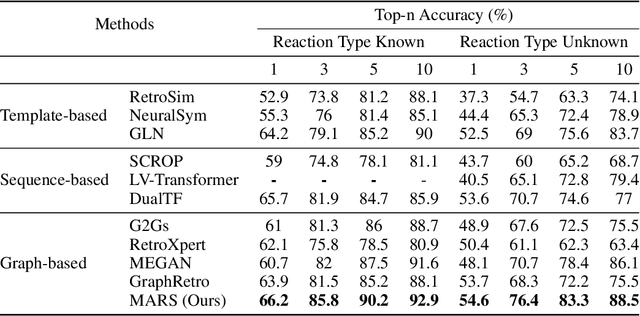
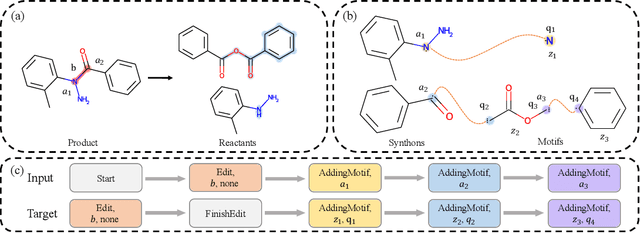
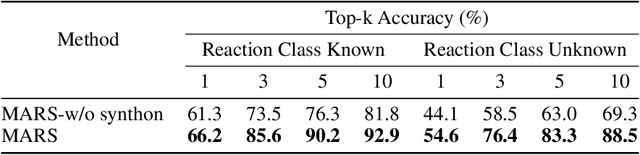
Retrosynthesis is a major task for drug discovery. It is formulated as a graph-generating problem by many existing approaches. Specifically, these methods firstly identify the reaction center, and break target molecule accordingly to generate synthons. Reactants are generated by either adding atoms sequentially to synthon graphs or directly adding proper leaving groups. However, both two strategies suffer since adding atoms results in a long prediction sequence which increases generation difficulty, while adding leaving groups can only consider the ones in the training set which results in poor generalization. In this paper, we propose a novel end-to-end graph generation model for retrosynthesis prediction, which sequentially identifies the reaction center, generates the synthons, and adds motifs to the synthons to generate reactants. Since chemically meaningful motifs are bigger than atoms and smaller than leaving groups, our method enjoys lower prediction complexity than adding atoms and better generalization than adding leaving groups. Experiments on a benchmark dataset show that the proposed model significantly outperforms previous state-of-the-art algorithms.