 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBuffer Matters: Unleashing the Power of Off-Policy Reinforcement Learning in Large Language Model Reasoning
Feb 24, 2026Traditional on-policy Reinforcement Learning with Verifiable Rewards (RLVR) frameworks suffer from experience waste and reward homogeneity, which directly hinders learning efficiency on difficult samples during large language models post-training. In this paper, we introduce Batch Adaptation Policy Optimization (BAPO), an off-policy RLVR framework to improve the data efficiency in large language models post-training. It dynamically selects training batches by re-evaluating historically difficult samples and reusing high-quality ones, while holding a lower bound guarantee for policy improvement. Extensive experiments further demonstrate that BAPO achieves an average 12.5% improvement over GRPO across mathematics, planning, and visual reasoning tasks. Crucially, BAPO successfully resolves 40.7% of problems that base models consistently fail to solve.
Fuz-RL: A Fuzzy-Guided Robust Framework for Safe Reinforcement Learning under Uncertainty
Feb 24, 2026Safe Reinforcement Learning (RL) is crucial for achieving high performance while ensuring safety in real-world applications. However, the complex interplay of multiple uncertainty sources in real environments poses significant challenges for interpretable risk assessment and robust decision-making. To address these challenges, we propose Fuz-RL, a fuzzy measure-guided robust framework for safe RL. Specifically, our framework develops a novel fuzzy Bellman operator for estimating robust value functions using Choquet integrals. Theoretically, we prove that solving the Fuz-RL problem (in Constrained Markov Decision Process (CMDP) form) is equivalent to solving distributionally robust safe RL problems (in robust CMDP form), effectively avoiding min-max optimization. Empirical analyses on safe-control-gym and safety-gymnasium scenarios demonstrate that Fuz-RL effectively integrates with existing safe RL baselines in a model-free manner, significantly improving both safety and control performance under various types of uncertainties in observation, action, and dynamics.
SrSv: Integrating Sequential Rollouts with Sequential Value Estimation for Multi-agent Reinforcement Learning
Mar 03, 2025Although multi-agent reinforcement learning (MARL) has shown its success across diverse domains, extending its application to large-scale real-world systems still faces significant challenges. Primarily, the high complexity of real-world environments exacerbates the credit assignment problem, substantially reducing training efficiency. Moreover, the variability of agent populations in large-scale scenarios necessitates scalable decision-making mechanisms. To address these challenges, we propose a novel framework: Sequential rollout with Sequential value estimation (SrSv). This framework aims to capture agent interdependence and provide a scalable solution for cooperative MARL. Specifically, SrSv leverages the autoregressive property of the Transformer model to handle varying populations through sequential action rollout. Furthermore, to capture the interdependence of policy distributions and value functions among multiple agents, we introduce an innovative sequential value estimation methodology and integrates the value approximation into an attention-based sequential model. We evaluate SrSv on three benchmarks: Multi-Agent MuJoCo, StarCraft Multi-Agent Challenge, and DubinsCars. Experimental results demonstrate that SrSv significantly outperforms baseline methods in terms of training efficiency without compromising convergence performance. Moreover, when implemented in a large-scale DubinsCar system with 1,024 agents, our framework surpasses existing benchmarks, highlighting the excellent scalability of SrSv.
SAMG: State-Action-Aware Offline-to-Online Reinforcement Learning with Offline Model Guidance
Oct 24, 2024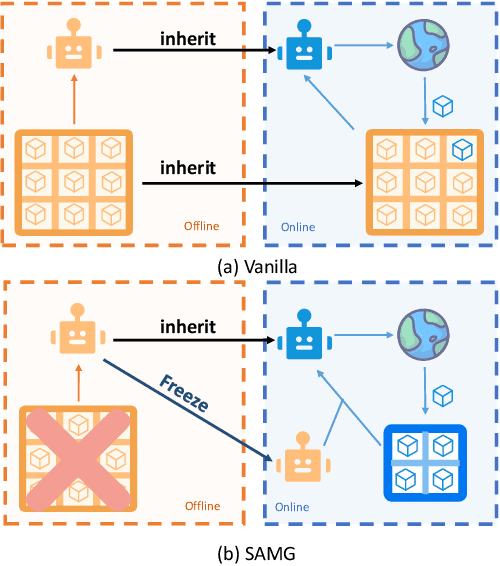
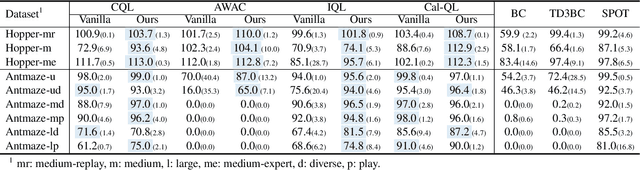
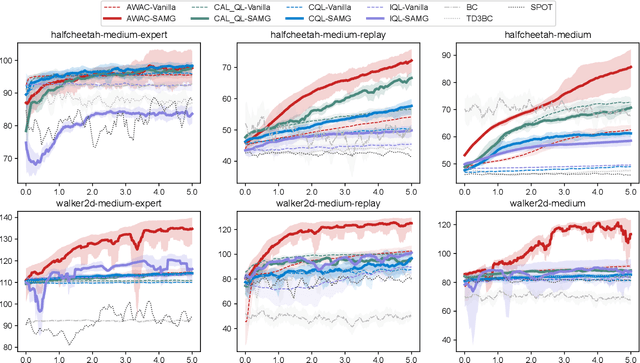
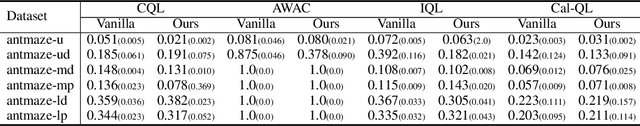
The offline-to-online (O2O) paradigm in reinforcement learning (RL) utilizes pre-trained models on offline datasets for subsequent online fine-tuning. However, conventional O2O RL algorithms typically require maintaining and retraining the large offline datasets to mitigate the effects of out-of-distribution (OOD) data, which limits their efficiency in exploiting online samples. To address this challenge, we introduce a new paradigm called SAMG: State-Action-Conditional Offline-to-Online Reinforcement Learning with Offline Model Guidance. In particular, rather than directly training on offline data, SAMG freezes the pre-trained offline critic to provide offline values for each state-action pair to deliver compact offline information. This framework eliminates the need for retraining with offline data by freezing and leveraging these values of the offline model. These are then incorporated with the online target critic using a Bellman equation weighted by a policy state-action-aware coefficient. This coefficient, derived from a conditional variational auto-encoder (C-VAE), aims to capture the reliability of the offline data on a state-action level. SAMG could be easily integrated with existing Q-function based O2O RL algorithms. Theoretical analysis shows good optimality and lower estimation error of SAMG. Empirical evaluations demonstrate that SAMG outperforms four state-of-the-art O2O RL algorithms in the D4RL benchmark.
ETDock: A Novel Equivariant Transformer for Protein-Ligand Docking
Oct 12, 2023



Predicting the docking between proteins and ligands is a crucial and challenging task for drug discovery. However, traditional docking methods mainly rely on scoring functions, and deep learning-based docking approaches usually neglect the 3D spatial information of proteins and ligands, as well as the graph-level features of ligands, which limits their performance. To address these limitations, we propose an equivariant transformer neural network for protein-ligand docking pose prediction. Our approach involves the fusion of ligand graph-level features by feature processing, followed by the learning of ligand and protein representations using our proposed TAMformer module. Additionally, we employ an iterative optimization approach based on the predicted distance matrix to generate refined ligand poses. The experimental results on real datasets show that our model can achieve state-of-the-art performance.
Predicting Protein-Ligand Binding Affinity with Equivariant Line Graph Network
Oct 27, 2022



Binding affinity prediction of three-dimensional (3D) protein ligand complexes is critical for drug repositioning and virtual drug screening. Existing approaches transform a 3D protein-ligand complex to a two-dimensional (2D) graph, and then use graph neural networks (GNNs) to predict its binding affinity. However, the node and edge features of the 2D graph are extracted based on invariant local coordinate systems of the 3D complex. As a result, the method can not fully learn the global information of the complex, such as, the physical symmetry and the topological information of bonds. To address these issues, we propose a novel Equivariant Line Graph Network (ELGN) for affinity prediction of 3D protein ligand complexes. The proposed ELGN firstly adds a super node to the 3D complex, and then builds a line graph based on the 3D complex. After that, ELGN uses a new E(3)-equivariant network layer to pass the messages between nodes and edges based on the global coordinate system of the 3D complex. Experimental results on two real datasets demonstrate the effectiveness of ELGN over several state-of-the-art baselines.