 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeZero-shot protein stability prediction by inverse folding models: a free energy interpretation
Jun 05, 2025Inverse folding models have proven to be highly effective zero-shot predictors of protein stability. Despite this success, the link between the amino acid preferences of an inverse folding model and the free-energy considerations underlying thermodynamic stability remains incompletely understood. A better understanding would be of interest not only from a theoretical perspective, but also potentially provide the basis for stronger zero-shot stability prediction. In this paper, we take steps to clarify the free-energy foundations of inverse folding models. Our derivation reveals the standard practice of likelihood ratios as a simplistic approximation and suggests several paths towards better estimates of the relative stability. We empirically assess these approaches and demonstrate that considerable gains in zero-shot performance can be achieved with fairly simple means.
A survey and benchmark of high-dimensional Bayesian optimization of discrete sequences
Jun 07, 2024Optimizing discrete black-box functions is key in several domains, e.g. protein engineering and drug design. Due to the lack of gradient information and the need for sample efficiency, Bayesian optimization is an ideal candidate for these tasks. Several methods for high-dimensional continuous and categorical Bayesian optimization have been proposed recently. However, our survey of the field reveals highly heterogeneous experimental set-ups across methods and technical barriers for the replicability and application of published algorithms to real-world tasks. To address these issues, we develop a unified framework to test a vast array of high-dimensional Bayesian optimization methods and a collection of standardized black-box functions representing real-world application domains in chemistry and biology. These two components of the benchmark are each supported by flexible, scalable, and easily extendable software libraries (poli and poli-baselines), allowing practitioners to readily incorporate new optimization objectives or discrete optimizers. Project website: https://machinelearninglifescience.github.io/hdbo_benchmark
A Continuous Relaxation for Discrete Bayesian Optimization
Apr 26, 2024To optimize efficiently over discrete data and with only few available target observations is a challenge in Bayesian optimization. We propose a continuous relaxation of the objective function and show that inference and optimization can be computationally tractable. We consider in particular the optimization domain where very few observations and strict budgets exist; motivated by optimizing protein sequences for expensive to evaluate bio-chemical properties. The advantages of our approach are two-fold: the problem is treated in the continuous setting, and available prior knowledge over sequences can be incorporated directly. More specifically, we utilize available and learned distributions over the problem domain for a weighting of the Hellinger distance which yields a covariance function. We show that the resulting acquisition function can be optimized with both continuous or discrete optimization algorithms and empirically assess our method on two bio-chemical sequence optimization tasks.
BEND: Benchmarking DNA Language Models on biologically meaningful tasks
Nov 25, 2023The genome sequence contains the blueprint for governing cellular processes. While the availability of genomes has vastly increased over the last decades, experimental annotation of the various functional, non-coding and regulatory elements encoded in the DNA sequence remains both expensive and challenging. This has sparked interest in unsupervised language modeling of genomic DNA, a paradigm that has seen great success for protein sequence data. Although various DNA language models have been proposed, evaluation tasks often differ between individual works, and might not fully recapitulate the fundamental challenges of genome annotation, including the length, scale and sparsity of the data. In this study, we introduce BEND, a Benchmark for DNA language models, featuring a collection of realistic and biologically meaningful downstream tasks defined on the human genome. We find that embeddings from current DNA LMs can approach performance of expert methods on some tasks, but only capture limited information about long-range features. BEND is available at https://github.com/frederikkemarin/BEND.
Internal-Coordinate Density Modelling of Protein Structure: Covariance Matters
Feb 27, 2023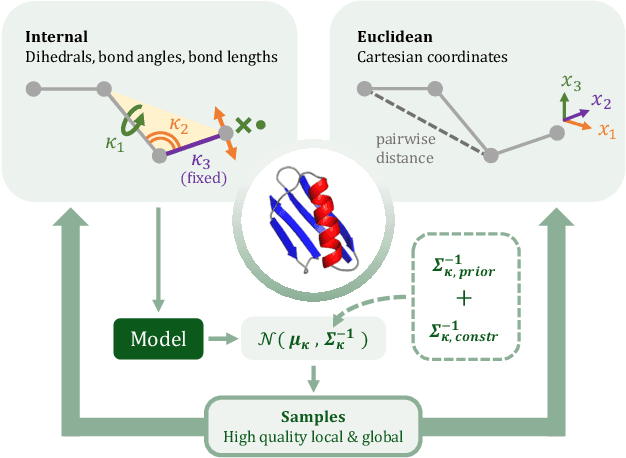
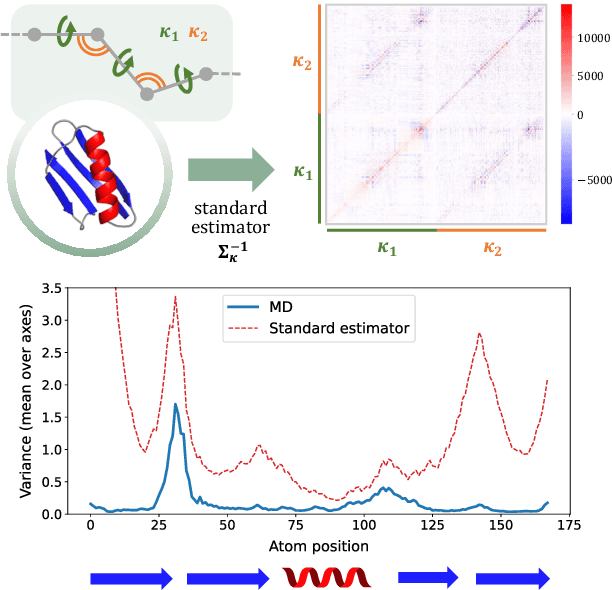
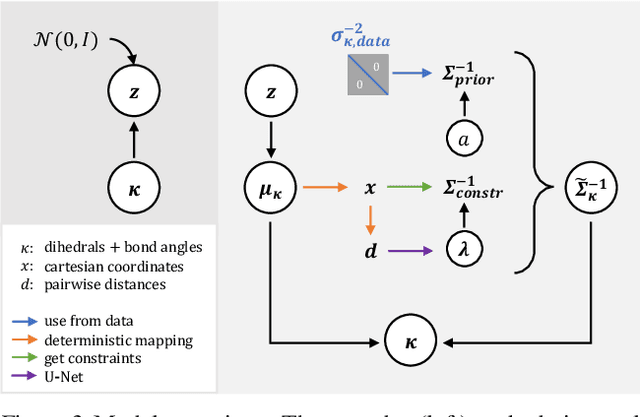
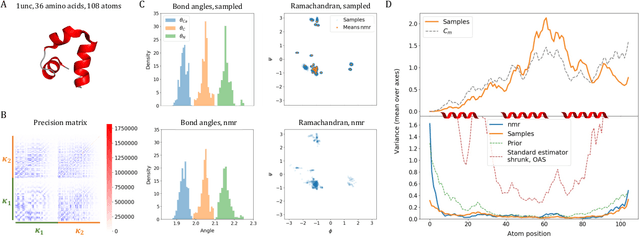
After the recent ground-breaking advances in protein structure prediction, one of the remaining challenges in protein machine learning is to reliably predict distributions of structural states. Parametric models of small-scale fluctuations are difficult to fit due to complex covariance structures between degrees of freedom in the protein chain, often causing models to either violate local or global structural constraints. In this paper, we present a new strategy for modelling protein densities in internal coordinates, which uses constraints in 3D space to induce covariance structure between the internal degrees of freedom. We illustrate the potential of the procedure by constructing a variational autoencoder with full covariance output induced by the constraints implied by the conditional mean in 3D, and demonstrate that our approach makes it possible to scale density models of internal coordinates to full-size proteins.
Adaptive Cholesky Gaussian Processes
Feb 23, 2022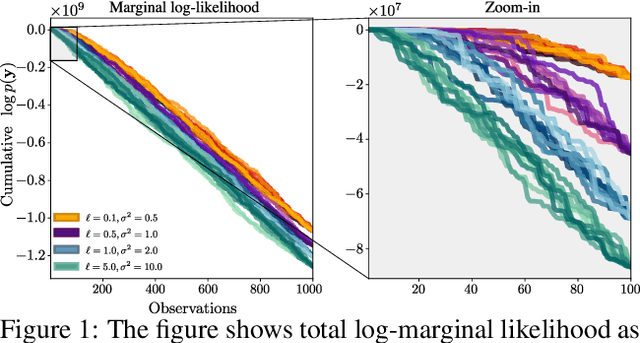
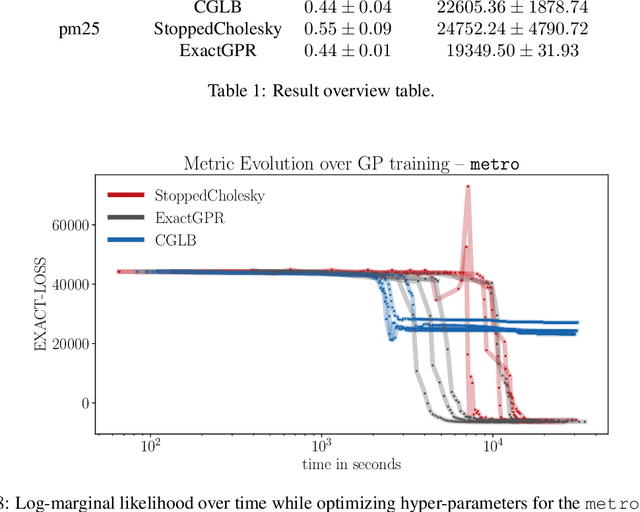
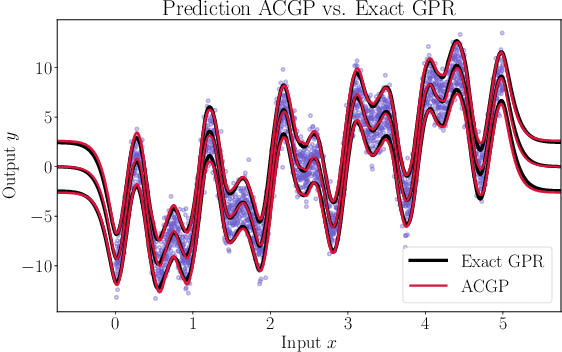
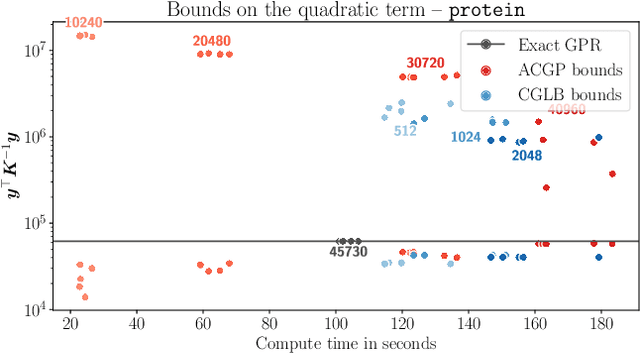
We present a method to fit exact Gaussian process models to large datasets by considering only a subset of the data. Our approach is novel in that the size of the subset is selected on the fly during exact inference with little computational overhead. From an empirical observation that the log-marginal likelihood often exhibits a linear trend once a sufficient subset of a dataset has been observed, we conclude that many large datasets contain redundant information that only slightly affects the posterior. Based on this, we provide probabilistic bounds on the full model evidence that can identify such subsets. Remarkably, these bounds are largely composed of terms that appear in intermediate steps of the standard Cholesky decomposition, allowing us to modify the algorithm to adaptively stop the decomposition once enough data have been observed. Empirically, we show that our method can be directly plugged into well-known inference schemes to fit exact Gaussian process models to large datasets.
What is a meaningful representation of protein sequences?
Nov 28, 2020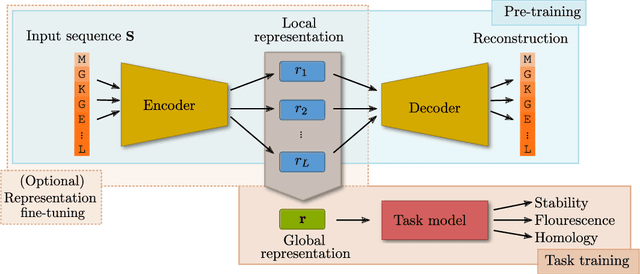
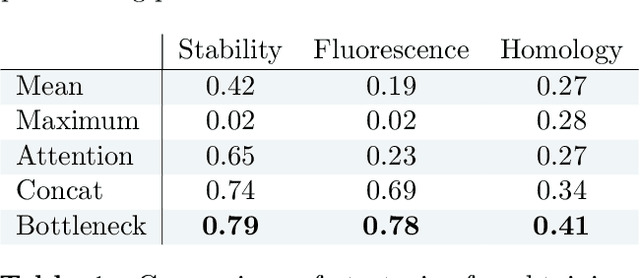
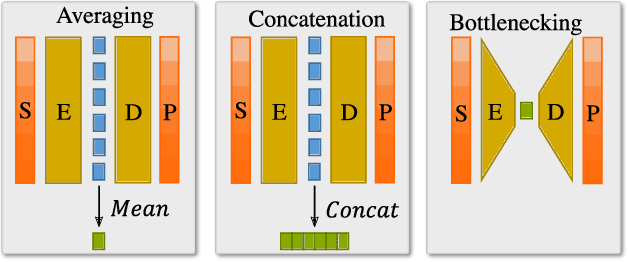
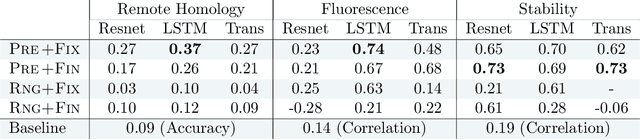
How we choose to represent our data has a fundamental impact on our ability to subsequently extract information from them. Machine learning promises to automatically determine efficient representations from large unstructured datasets, such as those arising in biology. However, empirical evidence suggests that seemingly minor changes to these machine learning models yield drastically different data representations that result in different biological interpretations of data. This begs the question of what even constitutes the most meaningful representation. Here, we approach this question for representations of protein sequences, which have received considerable attention in the recent literature. We explore two key contexts in which representations naturally arise: transfer learning and interpretable learning. In the first context, we demonstrate that several contemporary practices yield suboptimal performance, and in the latter we demonstrate that taking representation geometry into account significantly improves interpretability and lets the models reveal biological information that is otherwise obscured.
(q,p)-Wasserstein GANs: Comparing Ground Metrics for Wasserstein GANs
Feb 10, 2019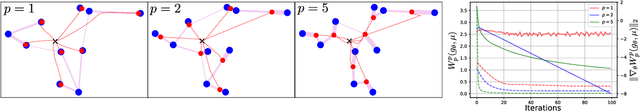
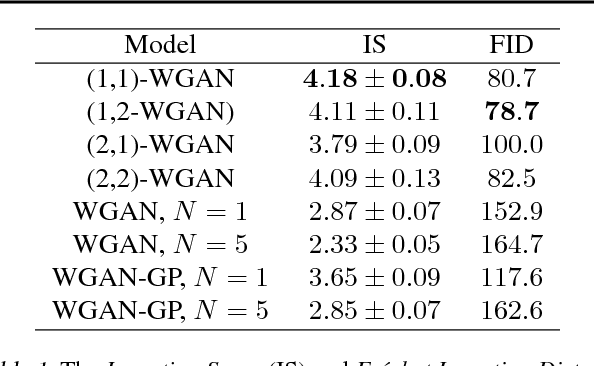
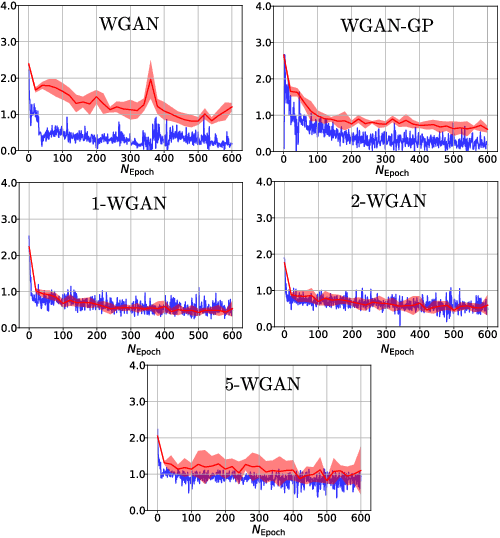
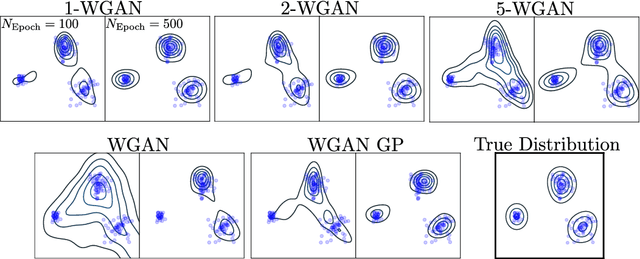
Generative Adversial Networks (GANs) have made a major impact in computer vision and machine learning as generative models. Wasserstein GANs (WGANs) brought Optimal Transport (OT) theory into GANs, by minimizing the $1$-Wasserstein distance between model and data distributions as their objective function. Since then, WGANs have gained considerable interest due to their stability and theoretical framework. We contribute to the WGAN literature by introducing the family of $(q,p)$-Wasserstein GANs, which allow the use of more general $p$-Wasserstein metrics for $p\geq 1$ in the GAN learning procedure. While the method is able to incorporate any cost function as the ground metric, we focus on studying the $l^q$ metrics for $q\geq 1$. This is a notable generalization as in the WGAN literature the OT distances are commonly based on the $l^2$ ground metric. We demonstrate the effect of different $p$-Wasserstein distances in two toy examples. Furthermore, we show that the ground metric does make a difference, by comparing different $(q,p)$ pairs on the MNIST and CIFAR-10 datasets. Our experiments demonstrate that changing the ground metric and $p$ can notably improve on the common $(q,p) = (2,1)$ case.
3D Steerable CNNs: Learning Rotationally Equivariant Features in Volumetric Data
Oct 27, 2018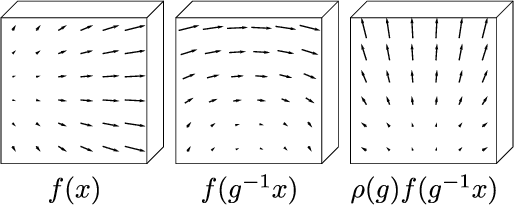

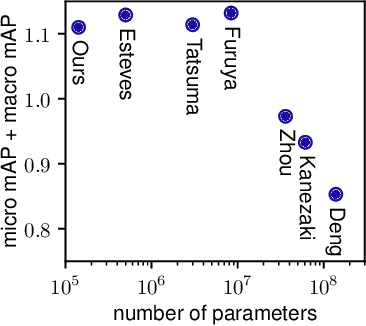
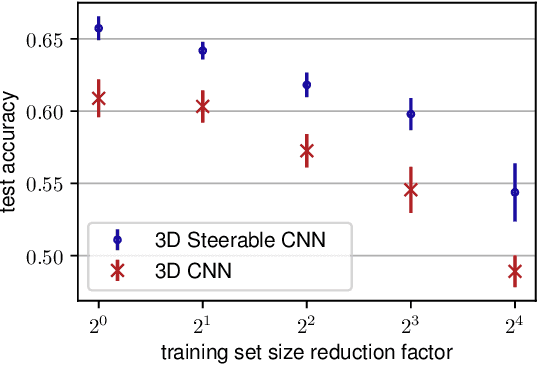
We present a convolutional network that is equivariant to rigid body motions. The model uses scalar-, vector-, and tensor fields over 3D Euclidean space to represent data, and equivariant convolutions to map between such representations. These SE(3)-equivariant convolutions utilize kernels which are parameterized as a linear combination of a complete steerable kernel basis, which is derived analytically in this paper. We prove that equivariant convolutions are the most general equivariant linear maps between fields over R^3. Our experimental results confirm the effectiveness of 3D Steerable CNNs for the problem of amino acid propensity prediction and protein structure classification, both of which have inherent SE(3) symmetry.